
Molecular Diagnostics Panels Test Information
We use custom, targeted, and reliable Next Generation Sequencing (NGS) chemistry to provide rule out testing for all genes in all of the gene panel tests at the sensitivity of Sanger sequencing and MLPA combined, including exon-level copy number assessment for all genes. All assays include confirmatory testing using Sanger sequencing, MLPA, q-PCR or others.
Molecular Diagnostics testing menu includes:
- Hereditary Cancer Panels (Ontario regional reference laboratory)
- Charcot-Marie-Tooth Gene Panel (Ontario reference laboratory)
- Mitochondrial Genome Sequencing (Ontario reference laboratory)
- Epilepsy Panels
- Lysosomal Storage Disorders (Ontario reference laboratory)
- Urea Cycle Disorders (Ontario reference laboratory)
- Adult Haemolytic Uremic Syndrome panel (available for research purposes only)
- Hematologic Oncology Molecular Testing
- Targeted Gene Testing
- Circulating Tumour DNA Testing
We are pleased to offer these services at competitive turn-around times (routine 4-6 weeks), at the industry leading cost efficiency.
“My clinic routinely uses LHSC molecular genetics laboratory for Charcot-Marie-Tooth Panel and Mitochondrial Genome sequencing which we previously obtained at various international laboratories. The service quality, responsiveness, reporting, and TAT meet and exceed international standards of quality at our own Canadian academic health care lab.”— Mark Tarnopolsky, MD, PhD, FRCP(C), Professor of Pediatrics and Medicine, President and CEO, Exerkine Corporation, Director of Neuromuscular and Neurometabolic Clinic, McMaster University Medical Center

Hereditary Cancer Panels (HCP)
Test Description:
The Hereditary Cancer Panel (HCP) is a genetic test designed to help assess the cancer predisposition risk for a number of common heritable cancers including: breast, ovarian, gastric, colorectal, pancreatic, melanoma, prostate, and endometrial cancers. The Hereditary Cancer Panel (HCP) includes 37 genes. Mutations in these genes are associated with clinically actionable results which will directly impact medical management recommendations and disease risk assessment.
HCP analysis will allow the assessment of an individual’s genetic test results which, in combination with their personal and family cancer history, will assist the clinician in determining an optimum pathway for their patient’s immediate medical and/or surgical management along with clinical follow up. Individuals identified as carrying highly penetrant, deleterious gene mutations, will be counseled on the potential effects of these mutations, and could be offered appropriate specialist medical or surgical referral for optimum ongoing management. For example, assessment of a patient at high risk of hereditary breast and/or ovarian cancer (HBOC) will take into account their personal and/or family cancer history, in conjunction with relevant gene analysis, to allow the attending physician to determine the optimum breast and/or gynecologic care for this individual. This could include high level surveillance options (e.g. breast MRI screening), and/or potential surgical intervention (mastectomy and/or hysterectomy/bilateral salpingo-oophorectomy) for individuals deemed to be at a significant level of risk.
Test Panels:
Hereditary Comprehensive Cancer Panel:AIP, APC, ATM, AXIN2, BAP1, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDC73, CDH1, CDK4, CDKN1B, CDKN2A, CHEK2, CTNNA1, DICER1, EGFR, EGLN1, EPCAM, EXT1, EXT2, FH, FLCN, GALNT12, GREM1, HOXB13, KIT, LZTR1, MAX, MEN1, MET, MITF, MLH1, MLH3, MSH2, MSH3, MSH6, MUTYH, NBN, NF1, NF2, NTHL1, PALB2, PDGFRA, PMS2, POLD1, POLE, POT1, PRKAR1A, PTCH1, PTEN, RAD51C, RAD51D, RB1, RECQL, RET, RNF43, RPS20, SDHA, SDHAF2, SDHB, SDHC, SDHD, SMAD4, SMARCA4, SMARCB1, SMARCE1, STK11, SUFU, TMEM127, TP53, TSC1, TSC2, VHL
Hereditary Breast/Ovarian/Prostate Panel: ATM, BARD1, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, EPCAM, HOXB13, MLH1, MSH2, MSH6, PALB2, PMS2, PTEN, RAD51C, RAD51D, STK11, TP53
Hereditary Endometrial Panel: BRCA1, BRCA2, EPCAM, MLH1, MSH2, MSH6, PMS2, POLD1, POLE, PTEN
Hereditary Gastrointestinal Panel: APC, ATM, BMPR1A, BRCA1, BRCA2, CDH1, CDKN2A, CHEK2, CTNNA1, EPCAM, GREM1, MLH1, MLH3, MSH2, MSH3, MSH6, MUTYH, NTHL1, PALB2, PMS2, POLD1, POLE, PTEN, SDHB, SDHD, SMAD4, STK11, TP53
Hereditary Lynch Syndrome Panel: EPCAM, MLH1, MSH2, MSH6, PMS2
Hereditary Gastric Panel: APC, ATM, BRCA1, BRCA2, CDH1, CTNNA1, EPCAM, MLH1, MSH2, MSH6, PALB2, PMS2, SDHB, SDHD, SMAD4, STK11, TP53
Hereditary Pancreatic Panel: ATM, BRCA1, BRCA2, CDKN2A, EPCAM, MLH1, MSH2, MSH6, PALB2, PMS2, STK11, TP53
Hereditary Polyposis Panel: APC, BMPR1A, EPCAM, GREM1, MLH1, MLH3, MSH2, MSH3, MSH6, MUTYH, NTHL1, PMS2, POLD1, POLE, PTEN, SMAD4, STK11, TP53
Familial Gastrointestinal Stromal Panel: KIT, PDGFRA, SDHA, SDHAF2, SDHB, SDHC, SDHD
Familial Melanoma Panel: BAP1, BRCA2, CDK4, CDKN2A, MITF, POT1, PTEN
Familial Renal Panel: BAP1, FH, FLCN, MET, MITF, PTEN, SDHA, SDHAF2, SDHB, SDHC, SDHD, TP53, TSC1, TSC2, VHL
Hereditary Pheochromocytoma/Paraganglioma Panel: FH, MAX, MEN1, NF1, RET, SDHA, SDHAF2, SDHB, SDHC, SDHD, TMEM127, VHL
Central Nervous System Cancer Panel: APC, EPCAM, LZTR1, MLH1, MSH2, MSH6, NF1, NF2, PMS2, POLE, POT1, PTCH1, PTEN, SMARCB1, SMARCE1, SUFU, TP53, TSC1, TSC2, VHL
Soft Tissue Cancer Panel: APC, ATM, BRCA1, BRCA2, CHEK2, EPCAM, MLH1, MSH2, MSH6, NF1, PMS2, TP53
Please see requisition for additional small panel orderables.
Test Indications:
Cancers suspected of being of hereditary origin frequently bear a number of hallmarks. Age at disease presentation is typically lower (under the age of 50) and it occurs in multiple family members. Rare cancers found in related family members are also indicative of a hereditary predisposition.
Epilepsy Panels
Test Description:
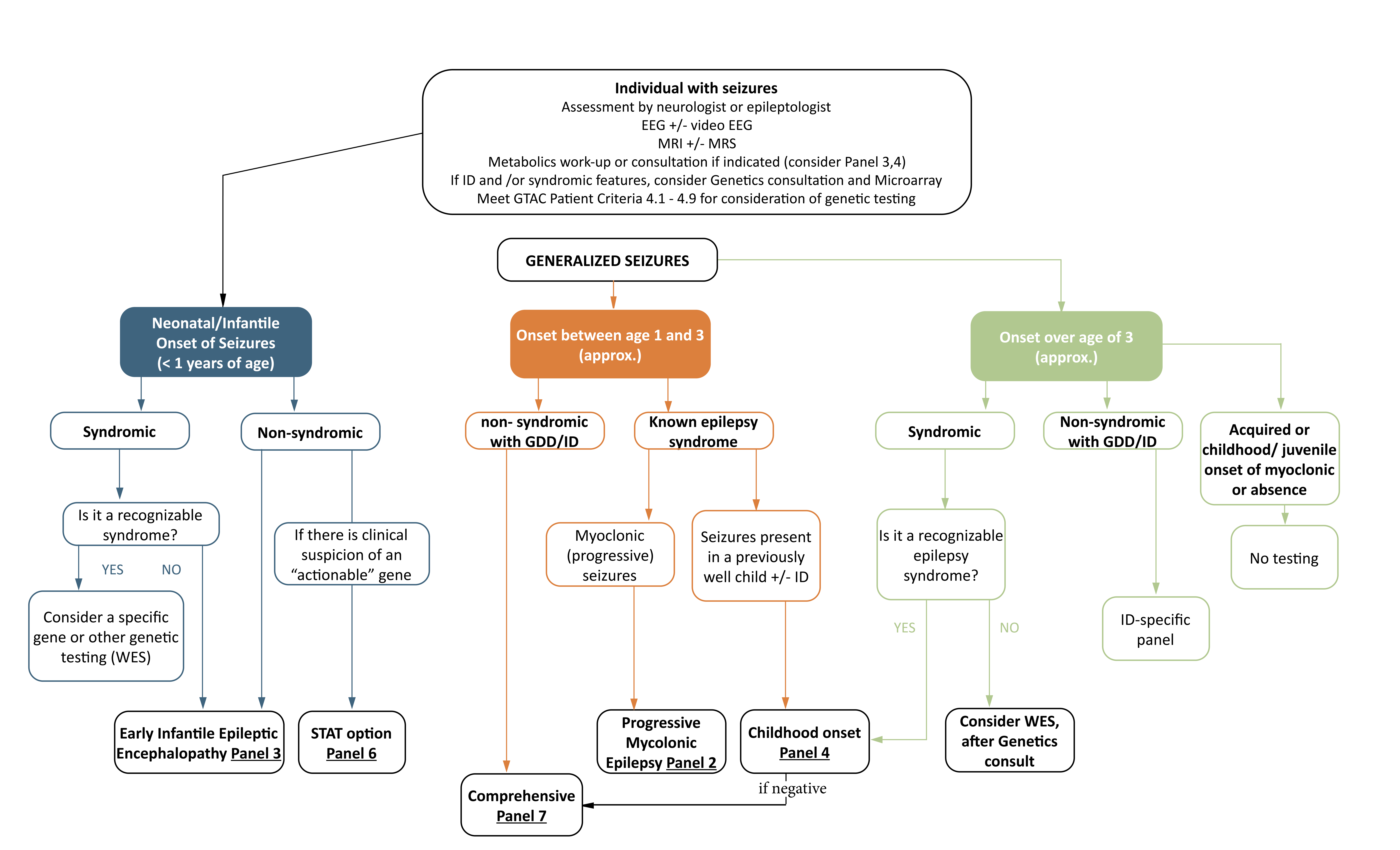
Epilepsy is a clinically heterogeneous disease with diverse aetiologies. Advances in molecular genetics over the last ten years have led to an explosion of novel genes implicated in monogenic and complex genetic epilepsies. Therefore, genetic testing now has become a critical part of the diagnostic evaluation of adults and children with epilepsy to identify genetic epilepsy syndromes, guide treatment, optimize genetic counseling, and bring closure and peace of mind to the families of those with a genetic disease whether treatable or not.
Based on the recommendations of an MOH epilepsy genetics advisory group, the Ontario Epilepsy Genetics Testing Program [OEGTP] provides evidence-based epilepsy gene panels�ID:31587668 for properly selected epilepsy patients. The latest revision of panel genes was performed in December 2023.
The test is designed to provide rule out level assessment for all coding sequence and copy number alterations for 190 genes, which encompass the majority of the known genetic etiologies for epilepsy. The test includes all major epilepsy syndromes and is subdivided into seven clinical categories:
- Comprehensive Epilepsy Panel (190 genes)
- Focal Epilepsy Panel (12 genes)
- Progressive Myoclonic Epilepsy Panel (21 genes)
- Early Infantile Epilepsy Panel (84 genes)
- Childhood Onset Epilepsy Panel (59 genes)
- Brain Malformation Epilepsy Panel (45 genes)
- Actionable Gene Epilepsy Panel (25 genes)
Please note that both sequence and copy number alterations (deletions/duplications) are routinely tested for every gene.
Test Panels:
Epilepsy Comprehensive panel: 190 Genes
ABAT, ACTB, ACTG1, ADGRG1, ADSL, AKT3, ALDH7A1, ALG13, AMT, AP3B2, ARFGEF2, ARHGEF9, ARV1, ARX, ASAH1, ASNS, ATP1A2, ATP1A3, ATP6V0A2, ATP7A, ATRX, B3GALNT2, CACNA1A, CACNA1E, CAD, CDKL5, CHD2, CHRNA4, CHRNB2, CLCN4, CLN3, CLN5, CLN6, CLN8, CNTNAP2, CSTB, CTSD, CTSF, DCX, DEPDC5, DNAJC5, DNM1, DOCK7, DYNC1H1, DYRK1A, EEF1A2, EHMT1, EPM2A, FGF12, FKRP, FKTN, FLNA, FOLR1, FOXG1, FRRS1L, GABBR2, GABRA1, GABRB2, GABRB3, GABRG2, GAMT, GLDC, GMPPB, GNAO1, GOSR2, GPSM2, GRIN1, GRIN2A, GRIN2B, GRIN2D, GRN, HCN1, HNRNPU, ITPA, KANSL1, KATNB1, KCNA1, KCNA2, KCNB1, KCNC1, KCNH5, KCNJ10, KCNMA1, KCNQ2, KCNQ3, KCNT1, KCTD7,KDM5C, KIF2A, LAMA2, LARGE1, LGI1, MBD5, MDH2, MECP2, MEF2C, MFSD8, MOCS1, NDE1, NEU1, NEXMIF, NGLY1, NHLRC1, NPRL2, NPRL3, NRXN1, OCLN, PAFAH1B1, PAK3, PCDH19, PHF6, PHGDH, PIGA, PIGG, PIGN, PIGO, PIGT, PIGV, PLCB1, PLPBP, PNKP, PNPO, POLG, POMGNT1, POMGNT2, POMK, POMT1, POMT2, PPT1, PRRT2, PSAT1, PSPH, PURA, RAB18, RAB39B, RAB3GAP1, RAB3GAP2, RELN, ROGDI, RTTN, SCARB2, SCN1A, SCN1B, SCN2A, SCN3A, SCN8A, SERPINI1, SGCE, SLC12A5, SLC13A5, SLC19A3, SLC25A12, SLC25A22, SLC2A1, SLC35A2, SLC6A1, SLC6A8, SLC9A6, SMARCA2, SNAP29, SPATA5, SPTAN1, SRD5A3, ST3GAL5, STX1B, STXBP1, SUOX, SYN1, SYNGAP1, SYNJ1, SZT2, TBC1D24, TCF4, TPP1, TRPM3, TSC1, TSC2, TUBA1A, TUBB, TUBB2A, TUBB2B, TUBB3, UBA5, UBE3A, VLDLR, WDR45, WDR62, WWOX, YWHAG, ZEB2
Focal Epilepsy panel: 12 Genes
CHRNA4, CHRNB2, DEPDC5, GRIN2A, KCNT1, LGI1, NPRL2, NPRL3, PRRT2, SCN1A, SCN1B, SLC2A1
Progressive Myoclonic Epilepsy panel: 21 Genes
ASAH1, CLN3, CLN5, CLN6, CLN8, CSTB, CTSD, CTSF, EPM2A, GOSR2, GRN, KCNC1, KCTD7, MFSD8, NEU1, NHLRC1, PPT1, SCARB2, SERPINI1, SGCE, TPP1
Early Infantile Epilepsy panel: 84 Genes
ABAT, ADSL, ALDH7A1, ALG13, AP3B2, ARHGEF9, ARV1, ARX, CACNA1A, CACNA1E, CAD, CDKL5, CHD2, DCX, DNM1, DOCK7, DYRK1A, EEF1A2, FGF12, FOLR1, FOXG1, FRRS1L, GABBR2, GABRA1, GABRB2, GABRB3, GABRG2, GAMT, GLDC, GNAO1, GRIN2A, GRIN2B, GRIN2D, HCN1, HNRNPU, ITPA, KCNA1, KCNA2, KCNB1, KCNH5, KCNQ2, KCNQ3, KCNT1, MDH2, MECP2, MEF2C, NGLY1, PCDH19, PIGA, PIGG, PIGN, PIGO, PIGT, PIGV, PLCB1, PNKP, PNPO, POLG, PRRT2, PURA, SCN1A, SCN1B, SCN2A,SCN8A, SLC12A5, SLC13A5, SLC25A12, SLC25A22, SLC2A1, SLC35A2, SLC6A8, SPATA5, SPTAN1, ST3GAL5, STX1B, STXBP1, SYNGAP1, SYNJ1, SZT2, TBC1D24, UBA5, WDR45, WWOX, YWHAG
Childhood Onset Epilepsy panel: 59 Genes
ADSL, ARX, ATP1A3, ATRX, CDKL5, CHD2, CLCN4, CNTNAP2, DEPDC5, DNAJC5, DYRK1A, EHMT1, FOXG1, GABBR2, GABRB2, GABRG2,GRIN2A, GRIN2D, KANSL1, KCNJ10, KCNMA1, KCNQ3, KDM5C, MBD5, MECP2, MEF2C, NEXMIF, NGLY1, NRXN1, PAK3, PCDH19, PHF6, PIGA, PIGN, PIGO, PNKP, POLG, PRRT2, RAB39B, ROGDI, SCN1A, SCN1B, SCN2A, SLC2A1, SLC6A1,SLC6A8, SLC9A6, SMARCA2, STX1B, SYN1, SYNGAP1, TBC1D24, TCF4, TRPM3, TSC1, TSC2, UBE3A, WDR45, ZEB2
Brain Malformation Epilepsy panel: 45 Genes
ACTB, ACTG1, ADGRG1, AKT3, ARFGEF2, ARX, ASNS, ATP6V0A2, B3GALNT2, B3GNT1(B4GAT1), DCX, DYNC1H1, FKRP, FKTN, FLNA, GMPPB, GPR56(ADGRG1), GPSM2, GTDC2(POMGNT2), KATNB1, KIAA1279(KIF1BP), KIF2A, LAMA2, LARGE, NDE1, OCLN, PAFAH1B1, POMGNT1, POMT1, POMT2, RAB18, RAB3GAP1, RAB3GAP2, RELN, RTTN, SGK196(POMK), SNAP29, SRD5A3, TUBA1A, TUBB, TUBB2A, TUBB2B, TUBB3, VLDLR, WDR62
Actionable Gene Epilepsy panel: 25 Genes
ALDH7A1, AMT, ATP7A, CAD, FOLR1, GAMT, GLDC, KCNQ2, KCNT1, MOCS1, PHGDH, PLPBP, PNPO, POLG, PSAT1, PSPH, SCN1A, SLC19A3, SLC2A1, SLC6A8, SUOX, TPP1, TRPM3, TSC1, TSC2
TEST INDICATIONS:
This Epilepsy panel test is a deep sequencing NGS assay designed as a rule out sequencing and copy number analysis test for all coding sequences of all genes tested. Content is designed by a panel of clinical experts Ontario MOHLTC Genetic Epilepsy Working Group to include majority of genes associated with epilepsy as the cardinal clinical presentation. In patients where epilepsy is not the cardinal clinical feature, and genetic etiology is suspected, other genetic and genomic analyses and clinical genetics referral may be considered.
Ontario Physicians must include completed questionnaire prior to submitting specimens to PaLM and confirm that age of onset, seizure type and electroclinical syndrome is consistent with a genetic epilepsy. Epilepsy Genetic Test Requistion
Test Indications:
The etiologies of genetic epilepsies are heterogeneous and a significant proportion of cases are attributable to structural brain defects and inherited metabolic disorders. Often it can be difficult to predict genotype based on electro-clinical phenotype. Individuals with epilepsy who may benefit from genetic testing include those with infantile onset, epilepsy refractory to treatment, epilepsy plus developmental delay, or families that may choose to have prenatal testing in future pregnancy. As this is an evolving field, the genotype-phenotype correlations are not well understood, and there is considerable phenotypic variability within the same genetic defect. For instance, mutations in the sodium channel genes cause Dravet syndrome as well as generalized epilepsy with febrile seizure plus. A particular electroclinical syndrome may be caused by multiple genetic defects. For example, Ohtahara syndrome may be caused by mutations in syntaxin binding protein-1 (STXBP1), potassium channel mutations (KCNQ2), Aristaless related homeobox (ARX), and solute carrier family 25, member 22 (SLC25A22) encoding a mitochondrial glutamate carrier.

CHARCOT-MARIE-TOOTH GENE PANELS
Test Description:
Charcot-Marie-Tooth (CMT) disease is a genetically and clinically heterogeneous group of inherited disorders of the peripheral nervous system characterized by progressive loss of muscle tissue and touch sensation across various parts of the body. CMT type 1 (CMT1) is a demyelinating peripheral neuropathy characterized by distal muscle weakness and atrophy, sensory loss, and slow nerve conduction velocity. CMT type 2 (CMT2) is an axonal (non-demyelinating) peripheral neuropathy characterized by distal muscle weakness and atrophy, mild sensory loss, and normal or near-normal nerve conduction velocities. CMT type 4 (CMT4) differ from CMT2 by autosomal recessive inheritance. Most common mutation is associated with CMT1A (70%-80% of all CMT1) and involves duplication of PMP22, while PMP22 gene deletion is the most common cause (80%) of hereditary neuropathy with liability to pressure palsies (HNPP).
Mutations in remaining genes are associated with less frequent subtypes of CMT, including autosomal dominant, recessive and X-linked forms of the disease.
Test Panels:
Charcot-Marie-Tooth panel-Comprehensive (87): AARS, ABHD12, AIFM1, ARHGEF10, ARHGEF28, ATP1A1, ATP7A, BAG3, BSCL2, C1orf194, CNTNAP1, DCTN1, DCTN2, DGAT2, DHTKD1, DNAJB2, DNM2, DNMT1, DRP2, DYNC1H1, EGR2, FBLN5, FGD4, FIG4, GARS, GDAP1, GJB1, GNB4, HARS, HINT1, HSPB1, HSPB3, HSPB8, IGHMBP2, INF2, JAG1, KARS, KIF1B, KIF5A, LITAF, LMNA, LRSAM1, MARS, MCM3AP, MFN2, MME, MORC2, MPV17, MPZ, MTMR2, NAGLU, NDRG1, NEFH, NEFL, PDK3, PDXK, PLEKHG5, PMP2, PMP22, PNKP, PRPS1, PRX, PTRH2, RAB7A, SBF1, SBF2, SCO2, SELRC1, SEPT9, SETX, SGPL1, SH3TC2, SIGMAR1, SLC12A6, SLC9A3R1, SORD, SPG11, SPTLC1, SURF1, TFG, TRIM2, TRPV4, TTR, VCP, VRK1, WARS, YARS
Test Indications:
CMT Type 1 and CMT X
- Nerve Conduction Study: Uniform conduction velocities in the upper limb <35 m/s
- Nerve Conduction Study: Intermediate conduction velocities 30-45 m/s, not necessarily uniform in distribution nor restricted to the upper limbs, and X-linked inheritance
Charcot-Marie-Tooth neuropathy type 1 (CMT1) is a demyelinating peripheral neuropathy characterized by distal muscle weakness and atrophy, sensory loss, and slow nerve conduction velocity. It is usually slowly progressive and often associated with pes cavus foot deformity and bilateral foot drop. Affected individuals usually become symptomatic between age five and 25 years. Fewer than 5% of individuals become wheelchair dependent. Life span is not shortened. CMT1A (70%-80% of all CMT1) involves duplication of PMP22. CMT1B (6%-10% of all CMT1) is associated with point mutations in MPZ. CMT1C (1%-2% of all CMT1) is associated with mutations in LITAF, and CMT1D (<2% of all CMT1) is associated with mutations in EGR2. CMT1E (<5% of all CMT1) is associated with point mutations in PMP22. CMT2E/1F (<5% of all CMT1) is associated with mutations in NEFL. Additional genes that can result in a demyelinating peripheral neuropathy include GDAP1 (CMT4A) and the X-linked GJB1 gene.
CMT Type 2
- Nerve Conduction Study: Uniform conduction velocities in the upper limb >40 m/s
Charcot-Marie-Tooth hereditary neuropathy type 2 (CMT2) is an axonal (non-demyelinating) peripheral neuropathy characterized by distal muscle weakness and atrophy, mild sensory loss, and normal or near-normal nerve conduction velocities. CMT2 is clinically similar to CMT1, although typically less severe. Peripheral nerves are not enlarged or hypertrophic. The subtypes of CMT2 are clinically similar and distinguished only by molecular genetic findings. The diagnosis is based on clinical findings and EMG/NCV characteristics. The genes in which mutations are known to cause CMT2 subtypes include MFN2 (CMT2A2), RAB7A (formerly RAB7) (CMT2B), LMNA (CMT2B1), TRPV4 (CMT2C), GARS (CMT2D), NEFL (CMT2E/1F), GDAP1 (CMT2H/K), X-linked GJB1, HSPB1 (CMT2F), MPZ (CMT2I/J), and HSPB8 (CMT2L). AARS(CMT2N), AIFM1 (CMTX4), DNAJB2 (CMT2T), DYNC1H1 (CMT2O), FGD4 (CMT4H), IGHMBP2(CMT2S), KIF1B (CMT2A1), LRSAM1 (CMT2P), MARS (CMT2U, MED25 (CMT2B2), MTMR2 (CMT4B1), NDRG1 (CMT4D), PDK3 (CMTX6), PRPS1 (CMTX5), SBF2 (CMT4B1), SPTLC1 (hSAN1A), TTR.
Hereditary neuropathy with liability to pressure palsies HNPP
Hereditary neuropathy with liability to pressure palsies (HNPP) presents as repeated focal pressure neuropathies including carpal tunnel syndrome and peroneal palsy with foot drop with onset in the second or third decade. HNPP usually presents with an autosomal dominant pattern of inheritance. Currently the only gene known to be associated with HNPP is PMP22. 80% of mutations detected are a deletion of the PMP22 gene region, and the remaining 20% are pathogenic variants in the PMP22 gene.

MITOCHONDRIAL GENOME SEQUENCING AND DEPLETIONS/INTEGRITY PANEL
Test Description:
The human mitochondrial DNA (mtDNA) encodes 37 genes coding for two rRNAs, 22 tRNAs and 13 polypeptides within its 16 569 bp. The mtDNA-encoded polypeptides are all subunits of enzyme complexes of the oxidative phosphorylation system. Disease phenotypes resulting from mitochondrial mutations may appear as distinct syndromes, such as Kearns-Sayre syndrome (KSS), Leber’s Hereditary Optic Neuropathy (LHON), mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS), chronic progressive external ophthalmoplegia (CPEO), myoclonic epilepsy with ragged-red fibers (MERRF), neurogenic weakness with ataxia and retinitis pigmentosa (NARP) or Leigh syndrome (LS). More frequently, the clinical presentation is much more heterogeneous. Some common symptoms include ptosis, external ophthalmoplegia, proximal myopathy, exercise intolerance, cardiomyopathy, sensorineural deafness, migraine, stroke-like episodes, pigmentary retinopathy, diabetes mellitus, encephalopathy, seizures, ataxia, and spasticity. This panel has been augmented with a selected series of 19 nuclear genes known to be associated the mitochondrial depletion disorders. About 80-95% of patients with mitochondrial disorders do not harbor a pathogenic mutation in the mitochondrial genome. A large proportion of these cases may have defects in nuclear-encoded genes that are involved in the biosynthesis of the mitochondrial genome or in the maintenance of mtDNA integrity. The Mitochondrial Genome Sequencing and Depletion/Integrity panel is appropriate for patients suspected of having one of the various forms of mtDNA depletion syndrome and/or mtDNA multiple deletions.
Test Panel:
Mitochondrial and Nuclear genes are a single panel
Mitochondrial Encoded:
MT-TY, MT-TW, MT-TV, MT-TT, MT-TS2, MT-TS1, MT-TR, MT-TQ, MT-TP, MT-TN, MT-TM, MT-TL2, MT-TL1, MT-TK, MT-TI, MT-TH, MT-TG, MT-TF, MT-TE, MT-TD, MT-TA, MT-RNR2, MT-RNR1, MT-ND6, MT-ND5, MT-ND4L ,MT-ND4 ,MT-ND3 ,MT-ND2 ,MT-ND1, MT-CYB, MT-CO3, MT-CO2, MT-CO1, MT-TC, MT-ATP8, MT-ATP6
Nuclear Encoded:
APTX, DGUOK, DNA2, FBXL4, GFER, MGME1, MPV17, OPA1, OPA3(isoformA & B), POLG, POLG2, RRM2B, SLC25A4, SPG7(isoform1 & 2), SUCLA2, SUCLG1, TK2, TWNK(C10orf2), TYMP

Hyperferritinemia Panel
(Research Use Only -- Awaiting Clinical Licensing)
Test Description:
Elevated serum ferritin concentrations are commonly found in the clinical arena. The cause of elevated ferritin concentrations is usually either increased ferritin synthesis or increased release of ferritin from damaged cells. Hyperferritinemia is common and often suggests the diagnosis of iron overload. However, many times it is elevated secondary to inflammation, obesity, alcohol use, or unknown causes. Most Caucasian patients with iron overload are homozygotes for the C282Y mutation of the HFE gene. There are a growing number of iron related genes that may contribute to an elevated ferritin and iron overload.
Our clinical NGS pipeline takes advantage of the custom targeted library design and patient batching resulting in the ability to analyse both sequence and copy number alterations in a single test. Utilization of this targeted NGS iron overload panel can be considered in addition to or as the replacement for the current HFE mutation screen.
Test Panel:
HFE2, STEAP3, SLC40A1, SLC25A38, TF, CP, HFE, TFR2, FTH1, CDAN1, B2M, HAMP, FTL, SEC23B, ALAS2
Test Indications:
This test is useful in the confirmation of a clinical diagnosis of hyperferritnemia and/or iron overload as part of a comprehensive clinical workup.

UREA cycle DISORDERS
Test Description:
Urea cycle disorders are a family of disorders in which excess ammonia found in the plasma is the result of either failure of ammonia detoxification or overproduction of ammonia. Defects in the urea cycle or its cofactors are not evident at birth, but symptoms frequently appear within days of birth. Cerebral edema, poor feeding and vomiting, seizures, hypothermia as well as hyper or hypoventilation. Primary hyperammonemia may be the result of an inherited deficiency of one of the enzymes composing the hepatic urea cycle and membrane transporters. Secondary hyperammonemia is the result of defects in the related pathways responsible for the assembly of ammonia acceptors and associated substrates required in the function of the urea cycle.
Heterogeneity in urea cycle disorders supports the use of a multi-gene panel for genetic testing. London Health Sciences Centre Urea Cycle Panel (LHSC UCD) is a Next Generation Sequencing (NGS) test involving sequence and copy number analysis of coding regions and adjacent intronic regions for 13 genes associated with various forms of urea cycle disorders.
The test is designed to provide rule out level assessment for all coding sequence and copy number alterations for 13 genes, which encompass the majority of the known genetic etiologies for urea cycle disorders. This test is useful in the confirmation of a clinical diagnosis, prenatal diagnosis in the presence of a family history of urea cycle disorders, as well as testing individuals with idiopathic hyperammonemia or suspicion of a urea cycle disorder.
Test Panel:
ARG1, ASL, CA5A, SLC25A13, ASS1, GLUL, CPS1, SLC25A15, SLC25A2, GLUD1, SLC7A7, OTC, CPS1
Test Indications:
This test is useful in the confirmation of a clinical diagnosis of urea cycle disorders, prenatal diagnosis in the presence of a family history of urea cycle disorders, as well as testing individuals with idiopathic hyperammonemia or suspicion of a urea cycle disorder. This information is may also be helpful when pursuing genetic counselling at risk family members. Individual genes may be selected when a known pathogenic variant previously identified in a family member can be used to determine the status of at risk pregnancies.

LYSOSOMAL STORAGE DISORDERS
Test Description:
Lysosomal storage disorders are a family of disorders in which there is a pathological accumulation of macromolecules in the lysosomes. This may be the result of defects in lysosomal enzyme function, or failure to transport across the lysosomal membrane. The accumulation of these macromolecules in turn result in cell damage and ultimately impaired organ function.
A list of more than 50 different inherited metabolic disorders fall under the category of lysosomal storage disorders.
The London Health Sciences Centre Lysosomal Storage Panel (LHSC LSD) is a Next Generation Sequencing (NGS) test involving sequence and copy number analysis of coding regions and adjacent intronic regions for 50 genes associated with various forms of lysosomal storage disorders. The test is designed to provide rule out level assessment for all coding sequence and copy number alterations for 50 genes, which encompass the majority of the known genetic etiologies for lysosomal storage disorders.
Test Panel:
AGA, ARSA, ARSB, ASAH1, CLN3, CLN5, CLN6, CLN8, CTNS, CTSA, CTSD, CTSK, DNAJC5, FUCA1, GAA, GALC, GALNS, GBA, GLA, GLB1, GM2A, GNPTAB, GNPTG, GNS, GRN, GUSB, HEXA, HEXB, HGSNAT, HYAL1, IDS, IDUA, LAMP2, LIPA, MAN2B1, MANBA, MCOLN1, MFSD8, NAGA, NAGLU, NEU1, NPC1, NPC2, PPT1, PSAP, SGSH, SLC17A5, SMPD1, SUMF1, TPP1
Test Indications:
This test is useful in the confirmation of a clinical diagnosis in conjunction with biochemical analysis and postnatal and prenatal diagnosis in the presence of a family history of lysosomal storage disorders.

ATYPICAL HEMOLYTIC UREMIC SYNDROME PANEL
Test Description:
Atypical hemolytic-uremic syndrome is a disease that primarily affects kidney function. This condition, which can occur at any age, causes abnormal blood clots (thrombi) to form in small blood vessels in the kidneys. These clots can cause serious medical problems if they restrict or block blood flow. Atypical hemolytic-uremic syndrome is characterized by three major features related to abnormal clotting: hemolytic anemia, thrombocytopenia, and kidney failure. Atypical hemolytic-uremic syndrome often results from a combination of environmental and genetic factors. Mutations in a number of genes can increase the risk of developing the disorder. PMID: 23251215; 24029428; 30294946
Test Panel:
Adult Onset Hemolytic Uremic Syndrome Gene Panel
C3, C9, CD46, CFB, CFH, CFHR1, CFHR2, CFHR3, CFHR4, CFHR5, CFI, DGKE, F12, FKRP, INF2, MMACHC, MMADHC, PLG, ST3GAL1, THBD, VWF

HEMATOLOGIC ONCOLOGY MOLECULAR TESTING
Test Description:
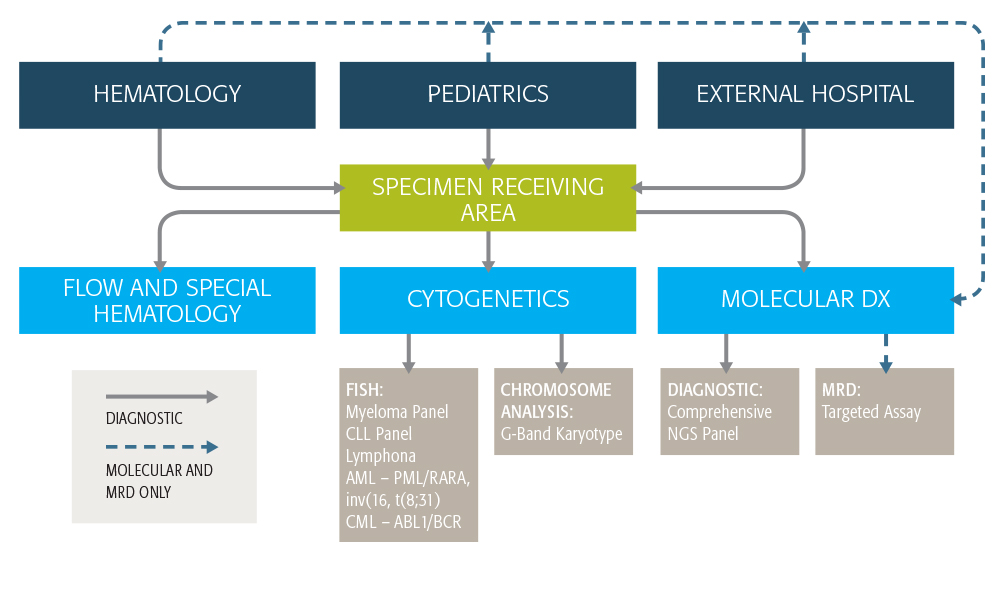
Hematologic Oncology NGS 40 gene (DNA) and 674 RNA (gene fusions)
Diagnostic assessment of patients with suspected hematologic malignancies follows a complex triaged protocol involving flow cytometric analysis, anatomic pathology, cytogenetics and molecular diagnostics. Classically, molecular genetics assessment is reserved for patients for whom initial assessment by flow cytometry, cytogentics or pathology indicates a possibility of a specific molecular subtype, followed by a targeted confirmatory molecular assay. This approach suffers from limitations including: inefficient coordination of complex triage procedures between different laboratories; insufficient specimens (often bone marrow) for repeat testing; increased turnaround times, and low diagnostic yield. It is only recently that scalable, high throughput, and high sensitivity-clinical-grade technology has become available enabling combined DNA sequencing and RNA-based assessment of fusion oncogenes. Now in routine clinical use, a NGS-based sequencing and gene fusion panel will be utilized for every patient specimen, in parallel to the standard karyotype assessment and flow cytometry. The panel includes assessment of 40 key DNA target genes, along with 29 driver genes involved in over 600 clinically-relevant gene fusions. Clinical validations highlighted an analytical sensitivity of 5% for detection of DNA sequence mutations (including small in/dels and more complex mutation such as FLT3 ITD) and 1% for detection of gene fusions. This approach offers considerably simplified molecular testing protocol (single common assay for all specimens), reduced TATs, and substantially increased molecular diagnostic yield in this patient population.
Test Panels:
DNA Mutation Hotspots sequencing (23): ABL1, BRAF, CBL, CSF3R, DNMT3A, FLT3, GATA2, HRAS, IDH1, IDH2, JAK2, KIT, KRAS, MPL, MYD88, NPM1, NRAS, PTPN11, SETBP1, SF3B1, SRSF2, U2AF1, WT1
Full genes sequencing (17): ASXL1, BCOR, CALR, CEBPA, ETV6, EZH2, IKZF1, NF1, PHF6, PRPF8, RB1, RUNX1, SH2B3, STAG2, TET2, TP53, ZRSR2
Gene fusions:: ABL1, ALK, BCL2, BRAF, CCND1, CRE, BBP, EGFR, ETV6, FGFR1, FGFR2, FUS, HMGA2, JAK2, KMT2A (MLL), MECOM, MET, MLLT10, MLLT3, MYBL1, MYH11, NTRK3, NUP214, PDGFRA, PDGFRB, RARA, RBM15, RUNX1, TCF3, TFE3
Note: Each of the above “driver” fusion genes may have multiple fusion “partners. This assay includes 674 of the most common fusion partner combinations of the above listed genes. For details please refer to: Thermo Fisher Ion AmpliSeq™ Oncomine Myeloid Assay.
Variant Assessment and Reporting:
Genetic variants are assessed and reported based on the Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer; A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists (PMID: 27993330). Tier I (level A and B) and Tier II (Level C and D) variant are reported; Tier III and Tier IV variants are not reported. Tier I Variants (variants of strong clinical significance, therapeutic, prognostic & diagnostic) include Level A (FDA/HC approved therapy; included in professional guidelines) and Level B (well powered studies with consensus from field experts). Tier II Variants (variants of potential clinical significance, therapeutic, prognostic & diagnostic) include Level C (FDA/HC approved therapy in another tumour type; investigational therapies; multiple small studies with some consensus) Level D (preclinical trials or few case reports without consensus). Tier III (variants of unknown clinical significance) and Tier IV (likely benign or benign) include low and high frequency variants, respectively, with no convincing published evidence of cancer association. Tier III and Tier IV variants are available upon request.
Targeted Molecular Assays:
| DNA ASSAYS (DIAGNOSTIC SPECIMENS) | |
|---|---|
| GENE | LIMIT OF DETECTION |
| NPM1 | 5% |
| CEBPA | 10% |
| FLT3-D835 | 1.25% |
| FLT3-ITD | 1.25% |
| JAK2 | 5% |
| CALR | 1.25% |
These DNA targeted assay are not used for MRD
| RNA ASSAYS (DIAGNOSTIC MRD MONITORING) | |
|---|---|
| GENE | LIMIT OF DETECTION |
| AML1/ETO (RUNX1/RUNX1T1) | Log 5 to 6 reduction |
| CBFB/MYH11 | Log 5 to 6 reduction |
| PML/RARA | Log 5 to 6 reduction |
| BCR/ABL-p190 and p210 | Log 5 to 6 reduction |
| TEL/AML1 (ETV6/RUNX1) | Log 5 to 6 reduction |
| TEL/AML1 (ETV6/RUNX1), | Log 5 to 6 reduction |
CLINICAL UTILITY:
| TEST | MUTATIONS | PROGNOSTIC IMPLICATION | TREATMENT IMPLICATION |
| AML | |||
| AML1/ETO (RUNX1-RUNX1T1) | Translocation t(8;21) | Favorable prognosis, M2 mostly, rarely: M1 or M4 | Induction followed by High Dose Cytarabine consolidations achieve long term remissions |
| CBFB/MYH11 inv | Inversion inv(16)(p13.1q22) | Favorable prognosis, intermediate when occurs with KIT mutation, nearly pathognomonic of M4eo-AML | Induction followed by High Dose Cytarabine consolidations achieve long term remissions.Association of c-kit has role for Allogeneic Stem Cell Transplant in CR1 |
| PML/RARA | Translocation t(15;17)(q22;q12) | Favorable prognosis, APML | Excellent Response to all-trans-retinoic acid and arsenic trioxide |
| BCR/ABL | Translocation t(9;22)(q34;q11) | M1 or M2 AML (3%), very poor prognosis | Tyrosine Kinase Inhibitors should be considered in chemotherapy regimens |
| FLT3 | ITD in exon 14 and 15, and D835 substitutions | With normal cyto poor to intermediate prognosis; with t(8;21) and inv(16) may be favorable | Response to FLT3 inhibitors with chemotherapy |
| NPM1 | Three most common insertion mutations (type A, B and D) (90-95%) | In AML, in absence of FLT3 and normal karyotype considered favorable prognosis | |
| CEBPA | Any loss-of-function mutations | AML WHO diagnostic category, favorable prognosis with norm karyotype / no other mutations, and both CEBPA alleles mutated | |
| ALL | |||
| TEL/AML1 (ETV6/RUNX1) | Translocation t(12;21)(p13;q22) | Favourable prognosis in Childhood ALL | Excellent response to L-Asparginase containing regimens |
| E2A/PBX1 (TCF3/PBX1) | Translocation t(11;19)(q23;p13) | Considered High Risk in Childhood ALL | |
| BCR/ABL-p190 | Translocation t(9;22)(q34;q11) | Seen in Adult as well as Childhood ALL | Inclusion of Tyrosine Kinase Inhibitors in chemotherapy regimens |
| MPN | |||
| JAK2 | V617F | Present in 97% of patients with PV and 50-60% of patients with ET or PMF | Response to JAK1/2 inhibitors irrespective of JAK2 mutation status |

Targeted Gene testing
Test Description:
A small group of inherited disorders are the result of predominantly single gene defects. A select group of diseases are overwhelmingly represented by a short list of specific variants (Cystic Fibrosis, LHON). A selection of these genes are available at the LHSC Molecular Genetics Laboratory.
See Test Menu for specific gene information.