All About Mitochondria
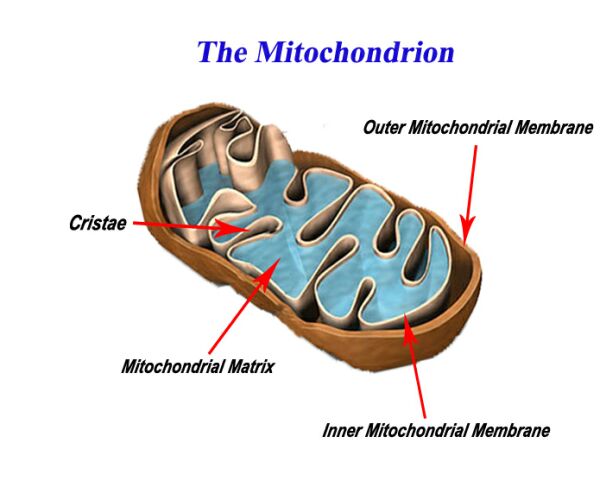
Inside the Mitochondrion
What are mitochondria?
Mitochondria are essential components of nearly all cells in the body. These organelles are the powerhouses for cells, providing energy to carry out biochemical reactions and other cellular processes. Mitochondria make energy for cells from the chemical energy stored in the food we eat.
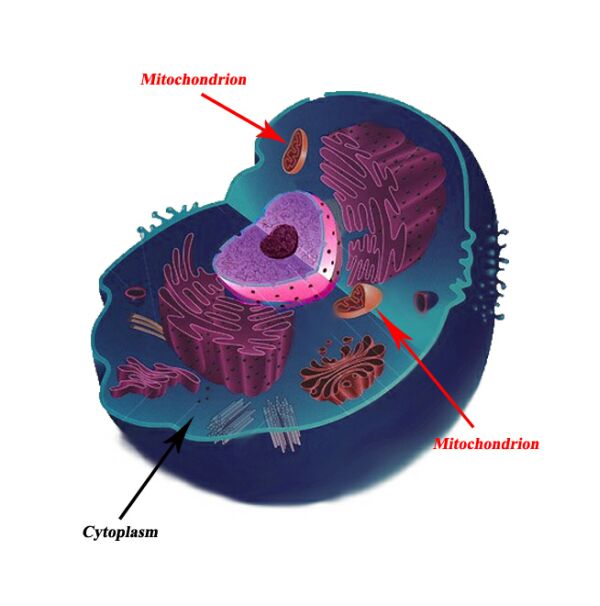
Where are mitochondria found?
Mitochondria are found in all body cells, with the exception of a few. There are usually multiple mitochondria found in one cell, depending upon the function of that type of cell. Mitochondria are located in the cytoplasm of cells along with other organelles of the cell.
History
How did mitochondria come about?
This question has been raised due to many characteristics shared by mitochondria and other single cellular living organisms. For example, mitochondria are the only organelles in the cell which contain their own DNA, as well as their own protein making machinery. Researched has shed light on the possibility of a theory known as endosymbiosis.
When life first began on our planet, single celled organisms produced energy in a way that was highly inefficient (anaerobic respiration, meaning without oxygen) compared to what most multi-cellular organisms use today (aerobic respiration, using oxygen). Through evolutionary time, plants came about and were able to produce oxygen in the atmosphere giving rise to aerobic respiration which produced energy in a highly efficient manner. The theory of endosymbiosis suggests that mitochondria were once free living organisms on their own that used aerobic respiration. Larger anaerobic cells simply engulfed these aerobic mitochondria to use their energy, giving rise to complex cells we find today such as those in our bodies.
Timeline of Mitochondrial Disease
The area of mitochondrial medicine is extremely new, and therefore ever expanding. The discovery of most mitochondrial diseases actually only occurred within the last 30 years. The timeline below shows some important milestones in the history of mitochondrial medicine:
1962 – The first case of suspected mitochondrial disease occurs where a woman has an extremely fast and efficient metabolism, and mitochondria that were larger in size and number in her muscle tissue
1962 – Chemical staining is applied to mitochondria, to identify any observable changes in mitochondria under a microscope
1975 – First case of MELAS is described
1981 – The mitochondrial genome is mapped
1982 – Scientific papers are published regarding Kearns-Sayre Syndrome and MERRF
1984 – First scientific paper published about MELAS
1991 – Biochemical and molecular analysis of tissue samples from patients becomes commercially available
How is energy made?
Our food contains the building blocks of life known as macromolecules, namely carbohydrates, proteins and fats. The energy stored in the molecular bonds of these molecules is converted into a usable energy source in the body known as ATP. ATP is the only energy currency that can be used in our bodies. This concept is analogous to energy from power plants entering our homes. Similar to macromolecules, there are many sources of energy including hydro, wind, nuclear etc. Although the sources are different, the energy that enters our homes is almost always converted to electricity to power various devices, similar to how only ATP in our cells is used to carry out cellular functions. The actual production of ATP is quite a complex process. The inner membrane of the mitochondrion is what is responsible for mass energy production.
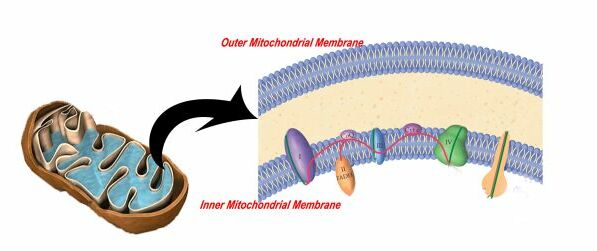
Specifically, five proteins form a chain on the inner mitochondrial membrane known as the respiratory chain that transfers energy (in the form of an electron) along these five proteins until it becomes ATP. This is shown in the animation below.
Basic Biology
The "Central Dogma of Biology"
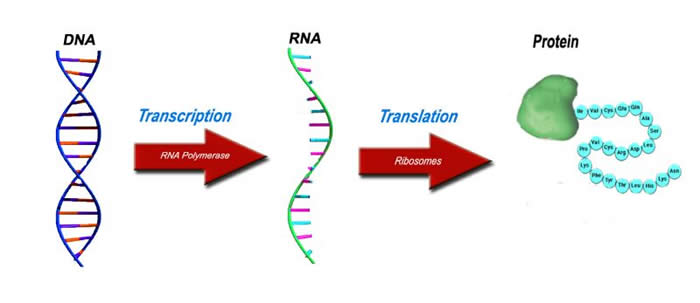
The idea of protein synthesis is often known as the central dogma since it the most elementary concept required to understand all of biology. All living things undergo the process of protein synthesis. The three major players in the central dogma are DNA, RNA and proteins.
The Original Blueprint - DNA
All living things require a blueprint, a recipe book, to make various essential molecules in our body namely proteins. Like most other organisms, the blueprint for humans is found in the form of DNA which we inherit from our parents. DNA is composed of four molecules known as bases: adenine, thymine, guanine and cytosine (A, T, G and C respectively). Segments of various sequences of these bases are what make up genes. Millions of such bases are found in a copy of DNA, allowing for an almost infinite number of combinations of bases to form genes.
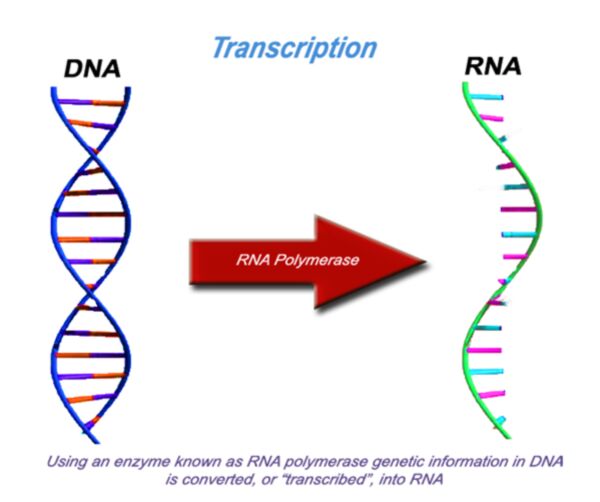
The modified photocopy - RNA
For proteins to be made, information stored in DNA must be first converted into another form. A process known as transcription converts the gene from DNA to RNA, a very similar molecule. This is like photocopying the original blueprint (DNA) onto a different type of paper. Through evolution, the protein making machinery known as the ribosome, can only understand genetic information in the form of RNA.
Depending upon what the gene codes for, the molecules may proceed onto becoming proteins or remain as RNA. The genes that remain as RNA and don’t proceed to become proteins, serve important functions for the cell including helping other RNA molecules in becoming proteins.
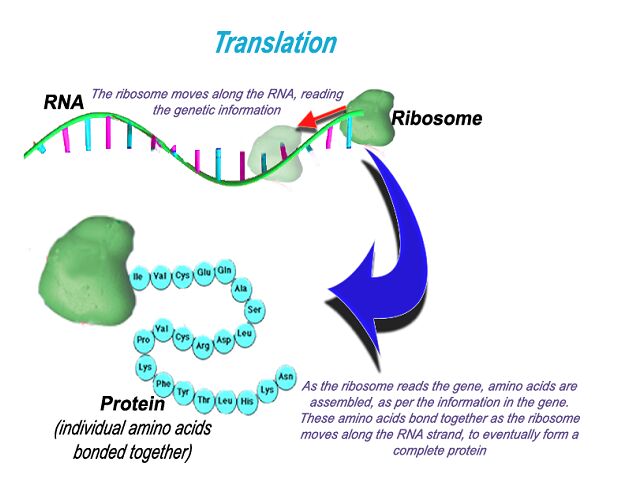
The Finished Product - Protein
Proteins are one of the macromolecules, which are essential building blocks of life. A protein is made up of many amino acids bonded together. They are quite abundant in the body, and serve various purposes. Enzymes acting at the molecular level, muscles that move our bodies, hair, nails and eye colour pigment are only a few examples of proteins found in the body.
Information stored in RNA is converted to proteins by a tiny organelle known as a ribosome. This protein making machine reads the sequence of bases in the RNA. This tells the ribosome where the gene starts, stops and what amino acids are required to assemble the protein. This protein is then transported within the cell where it is required. This central dogma of biology is observed in all living things.
General Symptoms
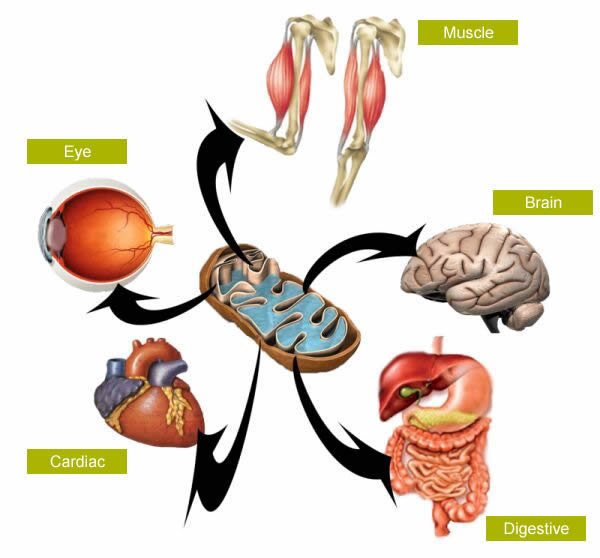
Mitochondrial diseases are extremely rare conditions with prevalence being approximately 12 in 100 000 people. Mitochondria are organelles found in nearly all body cells. Therefore, disorders of mitochondria are typically multisystemic, meaning they affect many organs and areas of the body.
Mitochondria are of course the energy powerhouses of the cell. A disease of these organelles disrupts their ability to make energy for the cell. This lack of energy to areas that demand it makes cells and eventually organs dysfunctional. This energy deficit can be considered the root cause of the symptoms observed in mitochondrial disease.
Neurological
Ataxia
- Meaning “without coordination”
- Uncoordinated movements of the arms and legs
Dementia
- A loss in intellectual ability
Migraine headaches
Myoclonus
- Twitching and jerking caused by sudden contraction of muscles
Peripheral neuropathy
- Damage or disease to nerves of the peripheral nervous system
- Numbness or tingling sensations in the extremities
Seizures
Sensorineural hearing loss
- Disease or damage of the nerve in the inner ear
- May cause complete deafness in one or both ears
Stroke
- Lack of oxygenated blood to the brain which may lead to paralysis, speech and vision problems, pain and other symptoms
Ocular
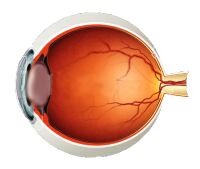
External ophthalmoplegia/ophthalmoperesis
- Paralysis/weakness of muscles in and around the eyes causing restricted movement of the eyes and appearance of drooping eyelids (ptosis)
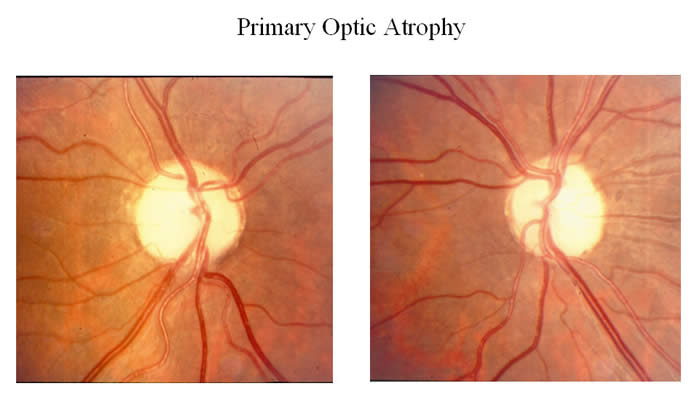
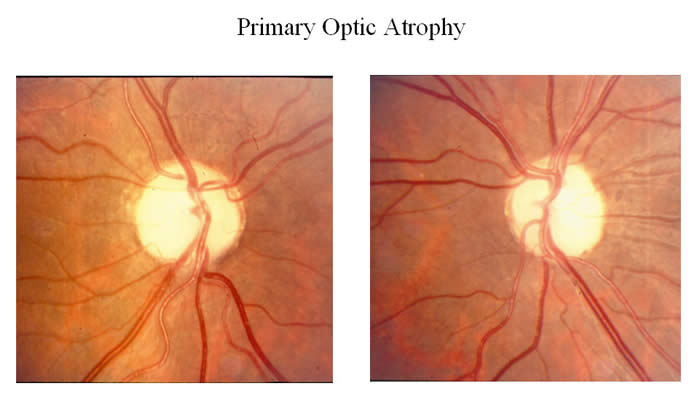
Optic neuropathy
- Damage to the optic nerve which connects photoreceptor cells to the brain, causing partial or complete blindness
Retinitis Pigmentosa – a group of diseases of the eye characterized by
- Swelling of cells in the retina
- Vision loss in the night
- Peripheral vision loss
- Central vision loss
Digestive
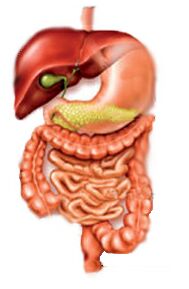
- Dysmotility (pseudo-obstruction)
- A group of disorders of the digestive tract caused by improper functioning of muscles that help move food down the tract
- This causes various symptoms of the digestive system, often caused by bacterial infections
Liver
- Hepatopathy
- Various forms of liver disease including jaundice, which causes a yellowish discolouration of the skin as a result of an excess chemical called bilirubin
Pancreas
- Diabetes mellitus
Muscular
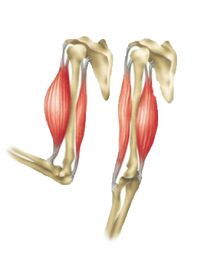
Exercise intolerance and fatigue
Fatigue
- Skeletal muscles that move our bodies require a lot of energy
- An energy deficit caused by a mitochondrial causes extreme weakness and constant fatigue
- Extreme muscle weakness also means physical activity becomes nearly impossible to tolerate
Myopathy
- A disease of muscle fibre, caused by insufficient energy supply to muscle cells
Cardiac
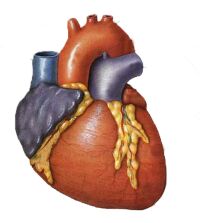
Cardiomyopathy
- A disease of the heart muscle which may lead to heart failure
Conduction Disorder
- Abnormalities in the conduction system that controls heartbeat
Diseases at the Molecular Level
Our DNA is made up of a sequence of “A”s “T”s, “G”s and “C”s known as bases. These four bases of DNA have specific pairing partners. An “A” always bonds with a “T” and a “G” always bonds with a “C”. When cells divide and DNA is replicated, there are regulatory checkpoints to ensure proper replication; however, mistakes may sometimes occur. A change in the blueprint (DNA), results in a changed product (protein). This altered protein structure may not function effectively which may be problematic for the mitochondria. After a certain threshold, the mitochondria as a whole may no longer be able to function and eventually lead to mitochondrial disease.
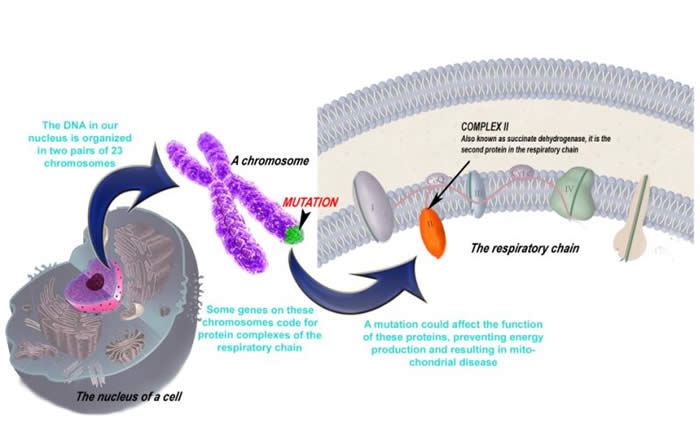
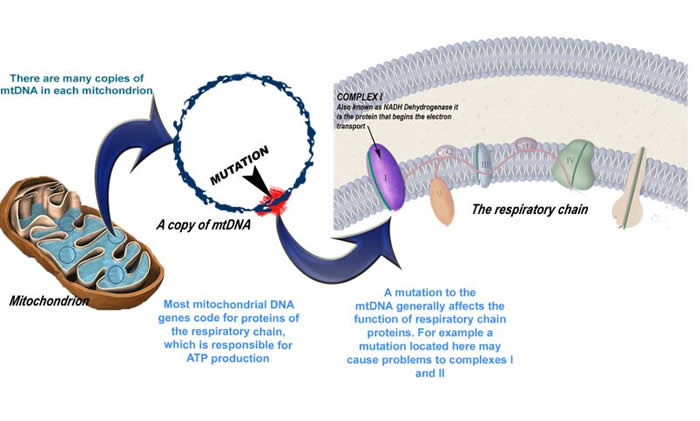
The images below show the effects of genetic changes at the molecular level
Nuclear Genes
Mitochondrial Genes
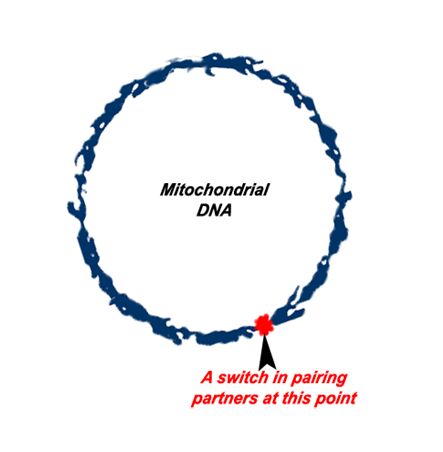
The Types of Mutations
1. A common replication error is the point mutation. The point mutation occurs when one these pairing partners switch (i.e. when a “G” pairs with an “A”). This slightly changes the resulting protein.
MELAS, MERRF, NARP, LHON, and Leigh's Disease
2. Another category of mutation is the deletion. Here, large scale portions of mitochondrial DNA are deleted, often due to environmental factors in the nucleus such as other genes or due to unknown causes. Large scale deletions means a loss in potentially vital mitochondrial genes, and therefore the effects can be fatal.

Kearns-Sayre Syndrome, CPEO, MNGIE, Alpers' Disease, and Pearson's Syndrome
Click here to view the concept of the above mutations in an animation
3. The third category of mitochondrial DNA mutations is the DNA depletions. Unlike our nucleus which only contains a single copy of our DNA, a single mitochondrion may contain tens of copies of its DNA. Like its namesake, DNA depletion diseases result in a reduction in the number of mitochondrial DNA copies.
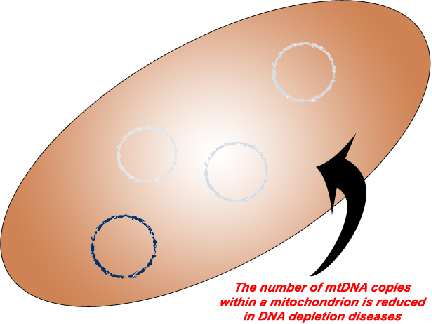
- SUCLA2 related DNA depletion syndrome
Inheritance Patterns
An individual may inherit a mitochondrial disease in one of many ways. How a disease is inherited, depends upon where the mutation exists. Wherever there is DNA, there is a potential source for a mutation. In a human cell there exist not one, but two full genomes. The first is the human genome located in the nucleus of the cell, where DNA is organized into structures called chromosomes. The second is mitochondrial DNA (mtDNA) which is located in the organelle after which it is named.
Nuclear DNA Diseases
What is the source of the problem?
Our DNA inside the nucleus is our entire body’s blueprint which we inherit from both of our parents. It is organized into two sets of 23 chromosomes (one set from each parent). Along the chromosomes are genes. An average human genome contains approximately 26 000 genes. A small fraction of those nuclear genes code for mitochondrial proteins, therefore a mutation in one of these genes could lead to mitochondrial disease.
If there is a mutation in a nuclear gene in my parents, what are the chances of me inheriting the mutation or the disease?
Recall that we have two copies of each chromosome—one from each parent. Both chromosomes contain the same genes and code for the same proteins, just slightly different versions of it. An example is eye colour; both parents have genes coding for eye colour yet the father may have the version for blue eyes and the mother may have the version coding for brown eyes. These different gene versions are called alleles. Variation exists within everyone’s genes—that is how we are all different. However, some genetic changes (or mutations) can cause the gene to not function properly and thereby cause disease. Genetic changes leading to disease are inherited in my different ways. The chance of being affected with a disease depends on many factors which are outlined below.
Autosomal Dominant
Like its namesake, autosomal dominant inheritance occurs because one allele “dominates” over the other. Of the two alleles, one genetic change on one allele will result in the condition being expressed, even if the other allele does not have a genetic change. This also means that at least one parent may also have the disease. The images below show possible scenarios:
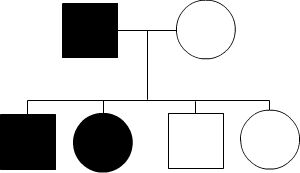
As seen above, having one affected parent who has both versions of the harmful allele, may give the disease to all their children. In contrast, a parent with only one version of the harmful allele may pass down the disease to only 50% of children.
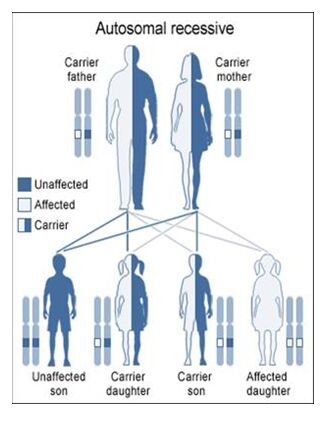
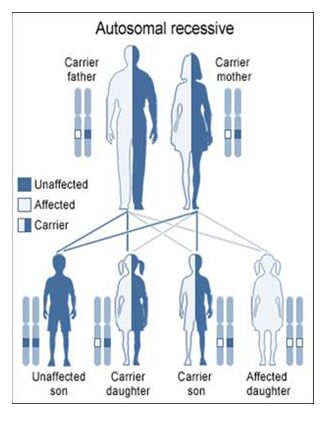
Autosomal Recessive
To acquire an autosomal recessive disease, both alleles in a person need to have a mutation. In autosomal recessive diseases the parents are known as carriers because only one of their genes has a mutation. Carriers are commonly asymptomatic (they do not show any signs of the disease), yet they carry a gene mutation that they can pass down to their children. It is common for the parents of a person with an autosomal recessive disease to be a carrier yet have no family history of the disease. The possible scenarios for inheriting autosomal recessive diseases are shown below:
The four scenarios in autosomal recessive diseases:
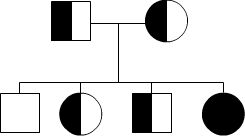
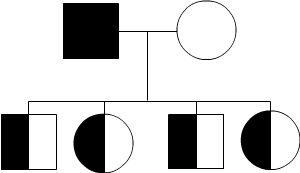
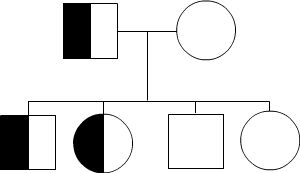
As seen above, it is only possible to inherit a recessive disease when a mutation exists on both sides of the family.
Mitochondrial DNA Diseases
Where is the genetic change?
As previously discussed, there are two sources of DNA in our body existing in two genomes—the nuclear DNA and mitochondrial DNA. Mitochondria are of course the energy producing organelles in the body.
Mitochondria have their own set of instructions, or blueprints—they have their own set of DNA. Mitochondrial DNA (mtDNA) contains genes that code for a number of proteins that are used by mitochondria (i.e. the proteins of the respiratory chain). Changes in mtDNA are another potential source of mitochondrial disease.
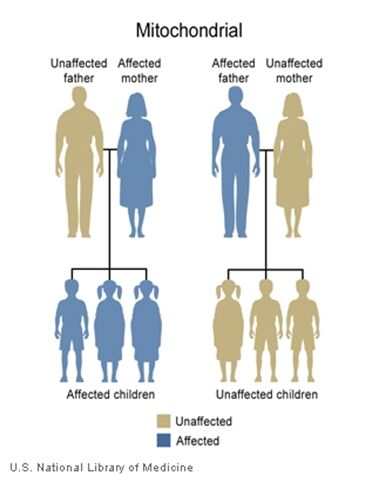
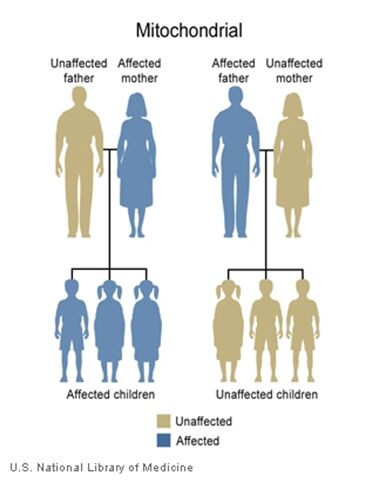
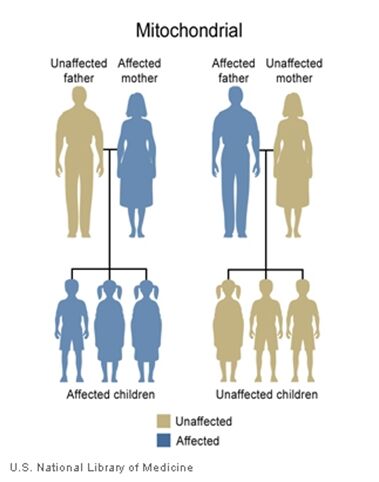
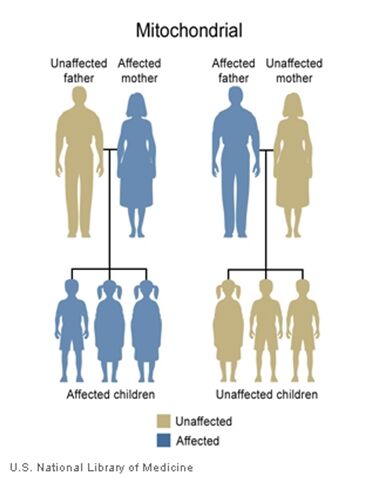
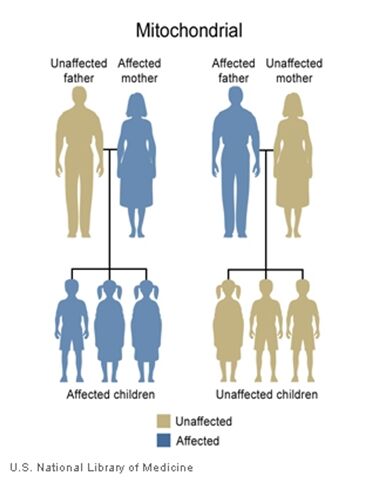
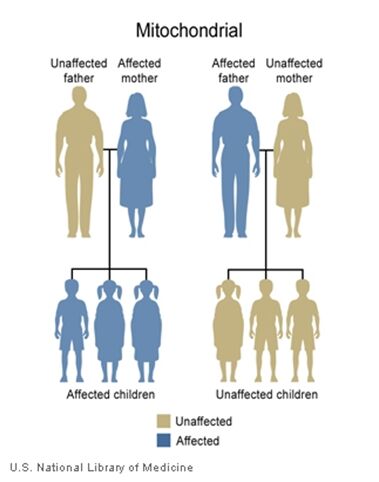
The inheritance of mtDNA is very different from the inheritance of nuclear DNA. It follows a pattern known as maternal inheritance. The process of conception and the relative contribution of materials from the sperm and the egg can help explain the concept. Sperm cells do not have mitochondria therefore MtDNA is not present in these cells, meaning the father will only contribute nuclear DNA and will not contribute any mtDNA. On the other hand, many mitochondria are present in a woman’s egg; therefore mtDNA from the mother is passed down to the next generation (in addition to nuclear DNA). Consequently if a mother has changes (mutations) in her mtDNA, these can be inherited by offspring. Essentially, mitochondrial diseases caused by mutations in the mtDNA can only be inherited from the mother, not the father.
The images of the pedigrees below illustrate this concept:
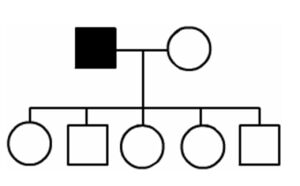
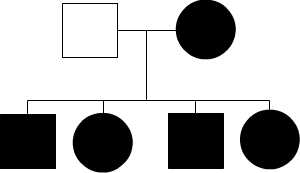
A father (left) would not pass down his mtDNA but a mother passes down all her mitochondrial traits to her children (right)
If there is a mutation in my parents’ mitochondrial DNA, what are the chances that I will inherit the mutation or be affected by the disease?
The answer to this question depends on which parent has a change in their mtDNA. If the mutation/disease is in the father, it will not be passed down. If it occurs in the mother, the mutation will almost always be passed down but the effects of the disease can be variable in the next generation. This occurs due to a phenomenon known as heteroplasmy.
As indicated above, mitochondria are present in a woman’s egg cell, as well as other cells in the body. At the conception of a child, the woman’s egg and the man’s sperm combine to form one complete genetic complement. Recall that all the mitochondria in this newly formed embryo are inherited from the mother.
All cells go through a process of cell division, including a developing embryo, which divides a cell into two distinct cells. During cell division, mitochondria are randomly dispersed throughout the cell. When a cell divides, the mitochondria present are divided randomly based on their positioning within the cell during division. If there is a mtDNA mutation present in any of the mitochondria, these can be divided into the newly formed cell. Based on the random nature of the dispersion of the mitochondria within the cell, there is sometimes an uneven distribution of mitochondria in these divided cells—some with mtDNA mutations and other without such mutations. If the patient has a disproportionate amount of mtDNA with mutations, the patient may surpass the threshold effect and express the disease.
Whether the next generation will be affected by the disease depends upon how mitochondria have divided themselves into the female’s ova.
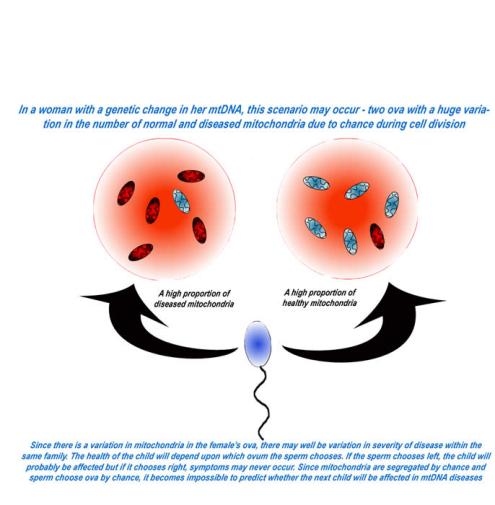
Each cell contains many mitochondria; likewise, each mitochondrion contains many copies of DNA. This leads to the concept of threshold effect. A certain number of mutations in the mtDNA must be present in any cell in order for symptoms to occur, at which time the “threshold” is overcome).
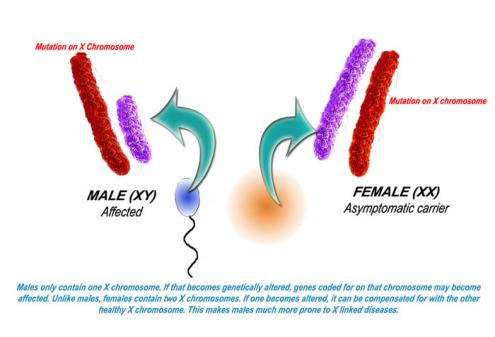
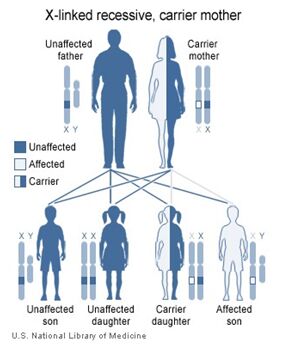
X-linked Inheritance
X-linked Inheritance involves the sex chromosomes, X and Y, which are present in the nuclear genome. Recall that a female has two X chromosomes, while males have one X and one Y. When there is a genetic change in one of the genes of the X chromosome the inheritance is said to be X-linked.
Males are more likely to express an X-linked condition because they have one X and one Y chromosome. When males inherit a genetic change on their X chromosome, they do not have a second copy of the gene; whereas females have two X chromosomes and therefore two copies of each gene. Therefore, if a female were to inherit a genetic change along her X chromosome, she would be a carrier for that associated condition. Females with a mutation present on both copies of a gene present on her X chromosomes would be affected with the associated disease.
Interestingly, there is a phenomenon known as “X-inactivation” or “Lyonization” that renders one copy of each of a female’s X-chromosomes inactive. This occurs early in the process of the development of the fetus. If the female’s X-inactivation is “skewed,” to include more inactive copies of the “normal” gene, then this female may exhibit some symptoms of the condition, while genetically she would be said to be a carrier. This phenomenon explains why some women who are carriers of an X-linked condition display symptoms of the disease, albeit typically less severe than their male counterparts.
Diagnoses of Mitochondrial Diseases
Multiple organ involvement and seemingly unrelated symptoms make the diagnosis of mitochondrial disorders extremely challenging. As a general rule of thumb, when three or more organ systems are involved without an evident cause, mitochondrial disease should be considered. On the contrary, if symptoms are not as severe then mitochondrial diseases may be overlooked and misdiagnosed.
1. Initial tests – body fluids: blood, urine and cerebrospinal fluid (CSF)
Basic biochemistry is the first test performed, with special attention given to the levels lactate and pyruvate. Often (but not always), mitochondrial disease patients suffer from a condition known as lactic acidosis which is an increase in lactic acid concentration in the body. Lactate and pyruvate are acids found in the mitochondria. Often in mitochondrial dysfunction, this ratio of lactate to pyurvate increases as a result of changes in biochemical pathways. When neurological symptoms are also present, this lactate to pyurvate ratio is tested for in the cerebrospinal fluid obtained from a lumbar puncture.
The levels of certain amino acids, proteins and organic acids are also measured in other body fluids such as urine.
Testing at the next stage is more symptomatic. Different diseases in different people present different symptoms. The commonly affected organs include (but are not restricted to) the brain, muscles, heart, eyes, the gastrointestinal tract, and the endocrine system.
2. Diagnostic tools
Neurological evaluation:
MRI and CT scans may show:
- Lesions (damage) to certain areas of the brain – symmetrical lesions to the area known as basal ganglia is common
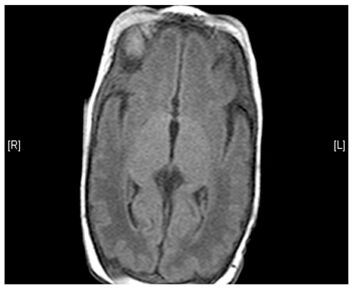
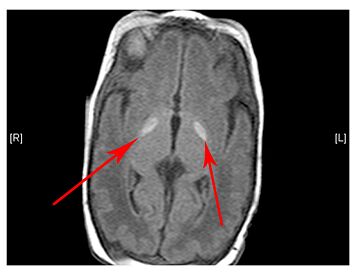
(Left) MRI scan of a healthy brain. (Right) The basal ganglia region of the brain shows abnormal signals.
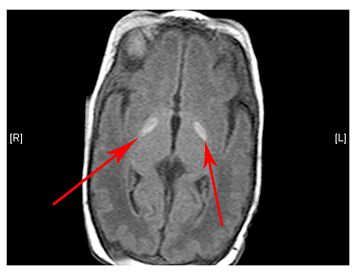
- Changes are observed in the grey matter and white matter areas in the brain
- Calcification (hardening) of certain areas, often the basal ganglia
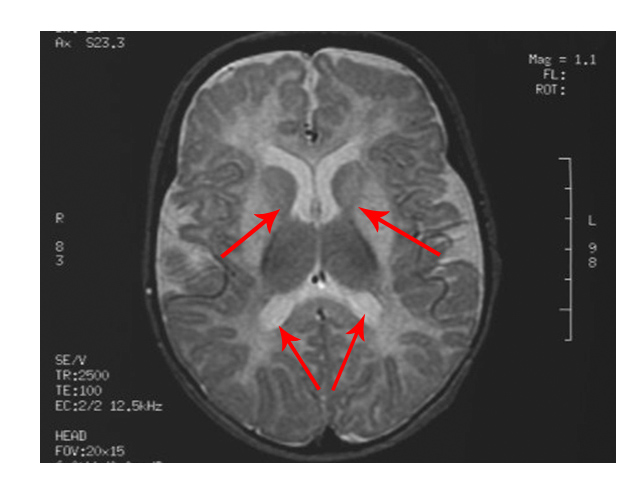
The scan shows changes in white matter within the brain as well as symmetrical abnormalities in signals (white areas) in the area known as the putamen (bottom arrows), in both hemispheres
- Atrophy (decrease in size) of certain brain areas
MRS tests may show
- Elevated levels of lactate in the brain
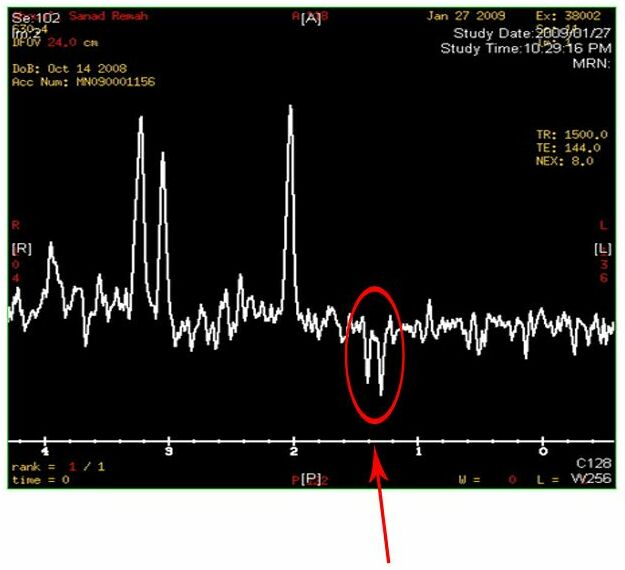
Two inverted peaks that resemble a “W” at around 1.2 on the horizontal axis indicate a high lactate concentration
- EEG may reveal a slow brain wave pattern associated with diseases of the nervous system
- Nerve conduction tests are useful in diagnosing myopathy and sensory neuropathy
Cardiac evaluation:
- Electrocardiography and echocardiography may reveal certain heart conditions such as a conduction disorder or cardiomyopathy
Ophthalmological evaluation:
- Electroretinography is helpful in diagnosing eye conditions such as retinitis pigmentosa
3. Finding the root cause - the levels of analysis
After step two, doctors generally have an idea as to which group of diseases they are looking at. Now the task is to medically classify it. The ultimate goal is to find the exact genetic change, or mutation, responsible for the observable symptoms. The order of analysis is at the tissue level, then protein level and finally gene level.
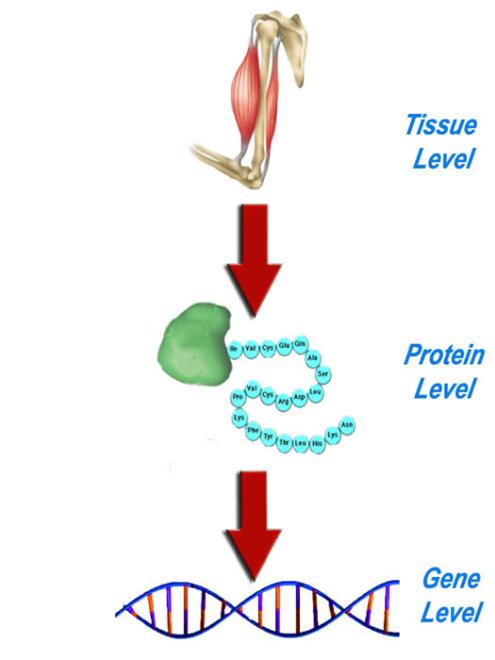
Tissue level – Biopsy
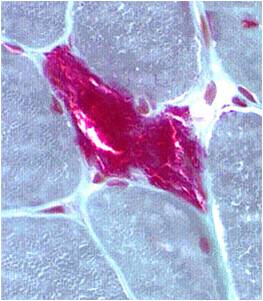
A muscle biopsy is most common when obtaining a tissue sample, primarily because it is the easiest to obtain. Depending upon the symptoms and disease however, other tissue samples such as from the skin or liver may be necessary. At this level of analysis, the histology (appearance) of the tissue is tested. Often observed are ragged red fibres (abnormal mitochondria beneath the muscle membrane appear as ragged red fibres when stained with a dye) which is characteristic of mitochondrial disease.
Ragged red fibres
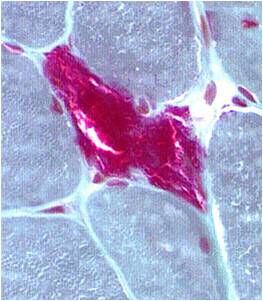
Chemical staining identifies diseased mitochondria, appearing as “ragged red fibres”. Left and right show lower and higher magnification, respectively.
Protein level – Enzymology
Mitochondrial disease is most frequently caused by dysfunctional proteins of the respiratory chain. Of course the respiratory chain is the pathway for energy production in mitochondria. Faulty proteins may severely reduce energy production in a cell thereby causing damage or cell death. To determine the cause of the disease, it is important to pinpoint the source of the problem. Enzymology tests measure the activity of the respiratory chain proteins. Chemical staining of these proteins is also used. These biochemical tests can exactly identify the faulty proteins in the chain.
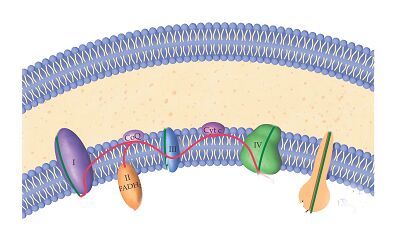
The function of each of the five proteins of the respiratory chain is tested using biochemical reactions to identify which proteins are faulty.
Gene level – DNA testing
The primary basis of any mitochondrial disease is at the level of DNA. There are numerous genes which code for proteins of the respiratory chain, found in both nuclear as well as mitochondrial DNA. Once the dysfunctional protein has been identified, the search begins to locate the genes responsible in coding for that protein complex. Using sophisticated methods such as sequencing the DNA one base at a time, we can determine the exact location of mutation. The type of mutation (deletion, duplication, point mutation etc) can also be identified. A specific type of mutation at a specific location is often the identity for a mitochondrial disease and distinguishes it from other diseases that may have similar characteristics.
Management of Diseases
Disclaimer: The following information is for education purposes only. The reader is advised to consult your physician regarding treatment issues. Despite a number of drugs that have been tried in mitochondrial disorders, there are no successful long term trials (Chinnery et al 2006).
Please consult your metabolic specialist regarding specific therapies for mitochondrial disorders.
Like many genetic diseases, the day to management patients with mitochondrial disorders remains a challenge. Although continuous research is being done, there is unfortunately no cure today for mitochondrial diseases. The primary goal is of symptom control and improvement in the quality of life as much as possible for patients. Unfortunately there are few randomized and controlled studies evaluating the various treatment options for mitochondrial disorders to develop clear and definite evidence based guidelines.
Mitochondrial disorders can manifest as single organ dysfunction or as multisystem disease involving the nervous system, eyes, ears, endocrine organs, heart, intestines, kidneys, bone marrow or skin etc.
Assessments
- Baseline: Complete medical assessment should be carried out.
- Vision and Hearing assessment (to rule out ophthalmological involvement) and hearing loss
- Cardiac assessment to rule out cardiomyopathy and arrhythmias
- Swallowing assessment
- Cognitive and developmental assessment
- Renal assessment
- Physiotherapy assessment
Therapeutic aspects for mitochondrial disorders
Even though there are presently no permanent cures that can be offered for mitochondrial disorders, the individual patient and family members who are affected can benefit from several therapeutic measures outlined below.
Symptomatic therapy
The symptomatic therapies can be divided into:
- Nutrition
- Drugs - (Symptomatic therapy)
- Drugs - affecting the mitochondrial function
- Avoidance of drugs
- Invasive measures
- Miscellaneous
- Future therapies
Nutrition
Though most specific diets have been found to be ineffective, overall general good nutrition is encouraged with avoidance of fasting. For some of the specific disorders such as pyruvate dehydrogenase deficiency, a ketogenic diet has been found to be useful. Some patients with MELAS and non specific mitochondrial disorders might benefit from relatively high-fat content in the diet.
If there are swallowing issues, a formal feeding and swallowing assessment should be carried out. Some patients may require gastrostomy or G Tube inserted directly into the stomach through the abdominal wall for feeding purposes. The procedure for insertion will usually require the services of a surgeon or interventional radiologist and a feeding team.
Drugs (Symptomatic therapy)
Antiepileptics: traditional anticonvulsant drugs are generally prescribed; however, special precautions should be taken to avoid the drug “valproic acid or sodium valproate, which can inhibit the carnitine uptake and may cause hyperammonemia.
Muscle relaxants: such as Baclofen or botulinum toxin can be considered for spasticity /dystonia (local or generalized) under supervision.
Pain: Gabapentin, Pregabalin, Carbamazepine or Lamotrigine can be used for neuropathic pain.
Cardiac drug therapy: is required in case of rhythm abnormalities or heart failure.
Gastrointestinal: Domperidone or cisapride may be useful if there is generalized dysmotility along with vomiting.
Renal: Electrolyte replacements (sodium and potassium) are required in when kidney function is compromised or in situations such as hypoaldosteronism where salt and fluid balance is affected.
Blood: In case of anemia or pancytopenia iron, transfusions or hematopoietic cell stimulators may be beneficial.
Endocrine: If there are endocrine disturbances along with mitochondrial disorders then patients may benefit from substitution of hormones for hypopituitarism, hypocorticism, hypogonadism etc.
Drugs-affecting the mitochondrial function
Respiratory chain proteins are responsible in the transference and electrochemical reactions which eventually lead to production of ATP, the body’s energy currency, as the end result. Vitamins help to make this transfer more efficient. The exact ingredients in the vitamin cocktail are determined by the physician and the success of the treatment varies from patient to patient. The important components of this cocktail include riboflavin, Coenzyme Q and vitamins E, C, etc.
Coenzyme Q (Quinones): This is most frequently prescribed supplement for mitochondrial disorders. Quinones not only have an antioxidant effect but also act as electron donor/acceptor respiratory chain. Coenzyme Q also known as coenzyme Q10, ubiquinone is a fat-soluble vitamin vital to a number of activities related to energy metabolism. CoQ is vital for the transfer of electrons from complex 1 and II to III. CoQ increases ATP production, has antioxidant properties and is an indirect stabilizer of calcium channels. Although uncommon, potential side effects of CoQ include gastrointestinal discomfort and low blood pressure. CoQ (300-1500 mg/day) is highly effective in CoQ deficiency states presenting as exercise intolerance and lactic acidosis. CoQ substitution and vitamin E have been effective in Friedrich ataxia as well.
Idebenone - Only limited experiences exist for idebenone
Other supplements
Vitamin E (Alpha-tocopherol) is usually used in combination with other supplements. There are no well-designed studies to evaluate its role on its own in mitochondrial disorders.
Alpha Lipoic acid is an essential cofactor for mitochondrial bioenergetics enzymes. It has been used in neuropathic pain in mitochondrial disorders with polyneuropathy.
Vitamin C acts as an important antioxidant and protects mitochondria from oxidative injury.
Riboflavin may be effective in secondary riboflavin deficiency. Riboflavin also has donor/acceptor properties. Riboflavin has been used in combination with other supplements. High dose riboflavin has been tried in headache and migraines with MELAS patients.
Succinate has been tried in some MELAS patients
Creatine-monohydrate Beneficial effect concerning muscle strength has been reported in some studies although not confirmed in others.
L- Arginine - Recent studies have shown that L-arginine , a nitric oxide precursor, may improve endothelial functions in patients with MELAS. L-arginine may be particularly beneficial in stroke- like events. Both intravenous administration (for acute episode) and oral arginine supplements for prevention have been tried with some success. Long term studies with use of arginine will be very helpful.
L-Carnitine - is highly effective in primary carnitine deficiency. Carnitine supplements are also helpful when there is secondary carnitine deficiency. L-Carnitine should also be given when valproate use is unavoidable.
Thiamine - Some patients with sideroblastic anemia may find thiamine useful. Patients with Pyruvate dehydrogenase deficiency may respond to thiamine.
Folic acid - has been found to be useful in Kearns Sayre syndrome (KSS) as CSF folic acid concentration is decreased.
Other vitamins such as Niacin, Pyridoxal phosphate, or vitamin K – only anecdotal reports are available.
Lactate lowering agents
Bicarbonate - Buffering of lactate is possible with bicarbonate but carries only a transient effect.
Dichloroacetate - decreases lactate by inhibiting pyruvate dehydrogenase enzyme complex. Side effects include peripheral neuropathy. Dichloroacetate does not reverse long term complications of lactic acidosis
Avoidance of Drugs
More important than administration is the avoidance of drugs which are detrimental to the mitochondrial function.
Special precautions should be taken to avoid the drug “valproic acid or sodium valproate", which can inhibit the carnitine uptake and may cause hyperammonemia.
Avoid drugs such as:
- ifosamide, carboplatin (inhibit mitochondrial replication)
- zidouvidine – reduces respiratory chain complexes 1/IV activity
- interferon (impair mitochondrial DNA transcription)
- carvedilol, bupivacaine (block respiratory chain 1 complex)
- acetyl salicylic acid (inhibit the electron transport chain)
- statins (decrease endogenous coenzyme Q)
- tetracyclins and amiodarone (inhibit beta oxidation)
- barbiturates, chloramphenicol (reduce mitochondrial protein synthesis)
- doxorubicin and valproate (decrease carnitine and reduce Respiratory chain/oxphos activity)
- metformin (biguanides) (cause lactic acidosis)
- avoid Propofol (anesthetic agent) (prolonged use can cause bradycardia, rhabdomyolysis, hyperlipidemia, metabolic and lactic acidosis)
DO NOT USE RINGERS LACTATE PRIOR TO OR DURING SURGICAL PROCEDURES
Invasive measures
- Blood transfusions for Pearson syndrome
- Use of erythropoietin and transfusion of platelets may be helpful in Pearson syndrome
- Implantation of pacemaker in patients with KSS syndrome.
- Implantable cardioverter defibrillator may be needed in case of hypertrophic cardiomyopathy.
- Ptosis has been corrected surgically although the effects of the repair may only be temporary.
- Cataracts due to mitochondrial disorders may require surgical removal.
- Cochlear implants may be helpful to overcome hearing problems.
- Pseudo obstruction in MNGIE or MELAS may require emergency surgical resection of part of intestine. Bone marrow transplantation has been tried as well for MNGIE.
- Gastrostomy (percutaneous) may be required for swallowing problems, recurrent vomiting and diarrhea.
Miscellaneous
Mitochondrial patients should avoid stress, extremes of temperature, alcohol, nicotine (Smoking).
They should get adequate sleep and should perform physical activity to below their maximum ability.
Future Therapies
Stem cell therapy has been used in patients with MNGIE. Gene therapy is challenging due to heteroplasmy. Manipulation of heteroplasmy levels (gene shifting). Shifting of the level of heteroplasmy towards wild type (normal) mitochondrial DNA is being pursued. This is being done by induction of muscle regeneration among other procedures. The easiest approach is exercise or endurance training which may improve muscle oxidative capacity.
Conclusion
With better understanding of the biology of mitochondrial function we hope that newer and more effective therapies will be developed. In the interim, it is important for patients to ensure proper nutrition, to avoid agents that can be harmful, to avoid smoking and alcohol. Exercise under proper supervision can be extremely helpful. Each patient should preferably have their own specific treatment protocol which can be used in event of emergencies. The patient and his/her family should get to know about their disease so that they can be active participants in their medical care. Genetic counselling is very important for the management. Prenatal diagnosis is still very challenging for mitochondrial disorders. A referral to an appropriate genetics clinic and an appointment with a geneticist and genetic counsellor can be very helpful.
Please see the mitochondrial support groups which may be of help.
References
Boehnke C, Reuter U, Flach U, Schuh-Hofer S, Einhaupl KM, Arnold G.
High-dose riboflavin treatment is efficacious in migraine prophylaxis: an open study in a tertiary care centre. Eur J Neurol. 2004 Jul;11(7):475-7.
Kubota M, Sakakihara Y, Mori M, Yamagata T, Momoi-Yoshida M.Beneficial effect of L-arginine for stroke-like episode in MELAS. Brain Dev. 2004 Oct;26(7):481-3; discussion 480.
Robert McFarland MRCPCH, Prof Robert W Taylor PhD and Prof Douglass M Turnbull FRCP, A neurological perspective on mitochondrial disease. The Lancet Neurology Volume 9, Issue 8, August 2010, Pages 829-840
Koga Y, Pvalko N, Nishioka J, Katayama K, Kakimoto N, Matsuishi T. MELAS and L-arginine therapy: pathophysiology of stroke-like episodes. Ann N Y Acad Sci. 2010 Jul;1201:104-10.
Kerr DS. Treatment of mitochondrial electron transport chain disorders: a review of clinical trials over the past decade. Mol Genet Metab. 2010 Mar;99(3):246-55. Epub 2009 Nov 26.
Shoffner JM, Wallace DC. Oxidative phosphorylation diseases and mitochondrial DNA mutations: diagnosis and treatment. Annu Rev Nutr. 1994;14:535-68.
Finsterer J. Treatment of mitochondrial disorders. European Journal of Paediatric Neurology 14. 2010 29-44.
P Chinnery, K Majamaa, D Turnbull and D Thorburn, Treatment for mitochondrial disorders, Cochrane Database Syst Rev 1 (2006) CD004426.
Online resources
http://www.ncbi.nlm.nih.gov/books/NBK1224/
Finsterer J. Treatment of Mitochondrial Disorders. Eur J Paediatr Neurol. 2010;
Disease Database
MELAS
What's in the name?
- MELAS - Mitochondrial Encephalomyopathy Lactic Acidosis and Stroke-like episodes is a rare inherited neurodegenerative disease
- Encephalo - Of, or relating to the brain/central nervous system (CNS)
- Myo-pathy - myo- Pertaining to muscles - pathy - a disease or disorder
- Lactic Acidosis - The condition of having lactic acid (in excess) in body fluids such as blood and cerebrospinal fluid
Who is affected?
- MELAS generally affects children, usually before adolescence
- The prevalence is only about 1 in 10 000
- There is no ethnic or gender predisposition to the disease
Symptoms
Clinical features:
- Short stature
- External ophthalmoplegia (a rare symptom in MELAS)
Paralysis of the muscles that open eyelids, and control eye movement
Results in drooping of the eyelids
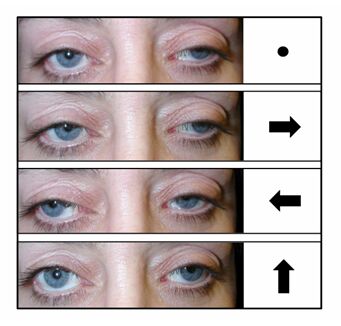
Neurological:
- Seizures
- Recurrent severe headaches and migraines
- Altered states of consciousness
- Dementia (loss of intellectual ability)
- Ataxia
From the Greek word meaning “without coordination”. Patients can report clumsiness in the movement of their hands, arms, legs as well as their balance.
- Temporary local paralysis
- Loss of sensation and strength in selective areas, following stroke like episodes
- Sensorineural hearing loss – damage to the nerve of the inner ear, resulting in deafness
Musculoskeletal:
- Overall muscle weakness
- Exercise intolerance
Other symptoms:
- Vomiting
- Loss of bowel control
- Kidney dysfunction
- Hormonal problems causing diseases like diabetes mellitus
- Cardiac conduction block – problems in the electrical system of the heart that controls heartbeat
Testing
Lab testing:
- Body fluids:
- A high lactic acid and pyruvic acid (acids found in the mitochondria) concentration in cerebrospinal fluid (CSF) and blood
- An increase in protein concentration in CSF
- Muscle Biopsy:
- Abnormal mitochondria from under the muscle tissue membrane appear as “ragged red fibres” under a microscope
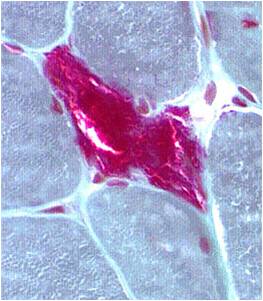
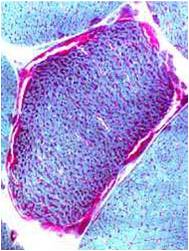
Chemical staining identifies diseased mitochondria, appearing as “ragged red fibres”. Left and right show lower and higher magnification, respectively.
Brain scans using MRI or CT reveal:
- Abnormal signals from certain brain areas, often from the basal ganglia
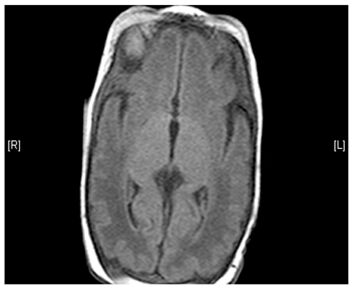
(Left) MRI scan of a healthy brain. (Right) The basal ganglia region of the brain shows abnormal signals.
- Damage to certain brain areas following stroke
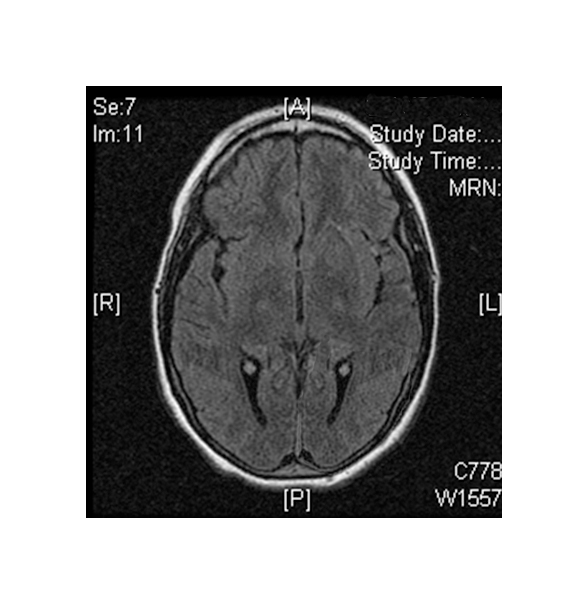
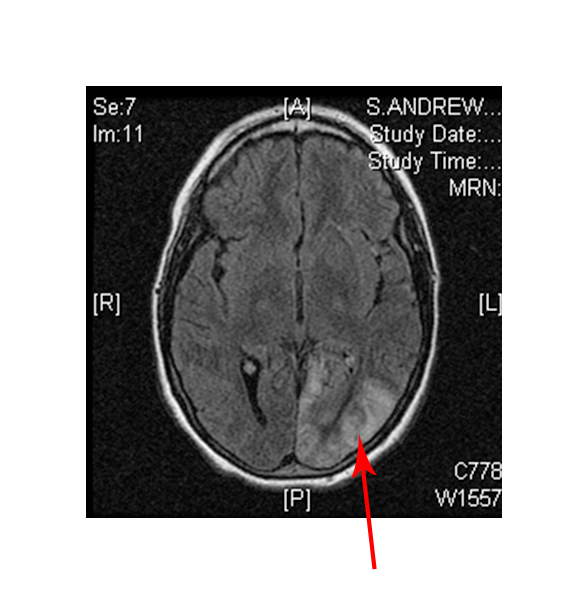
(Left) MRI scan of a healthy brain. (Right) Damage to the brain, after a MELAS patient suffered a stroke.
- Changes in the area of the brain known as white matter
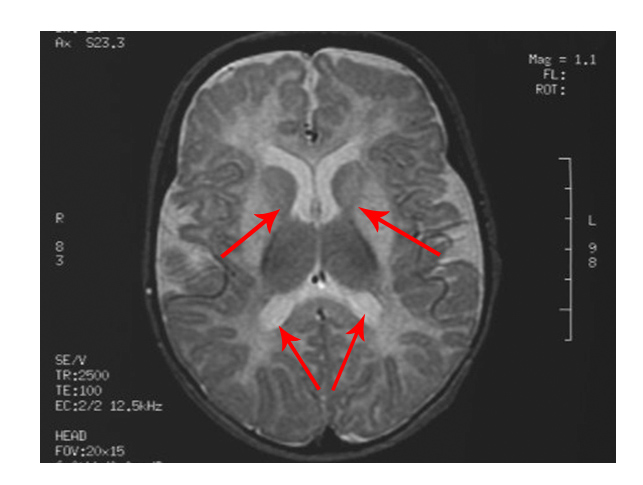
The scan shows changes in white matter within the brain as well as symmetrical abnormalities in signals (white areas) in the area known as the putamen (bottom arrows), in both hemispheres
- MRS testing reveals increase in lactate concentration in the brain
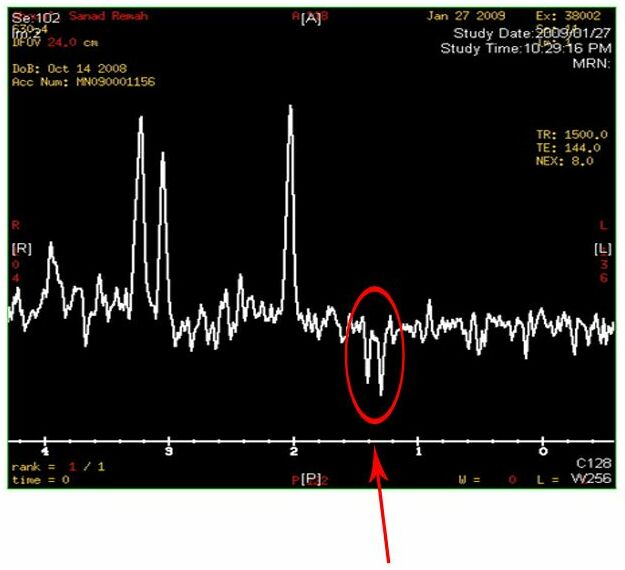
Two inverted peaks that resemble a “W” at around 1.2 on the horizontal axis indicate a high lactate concentration
- Electrocardiogram (ECG) tests may reveal problems with the cardiac muscle, or problems with the conduction system of the heart
- Testing enzymes of the respiratory chain often reveal defects in particular proteins – complex I and IV are common in MELAS
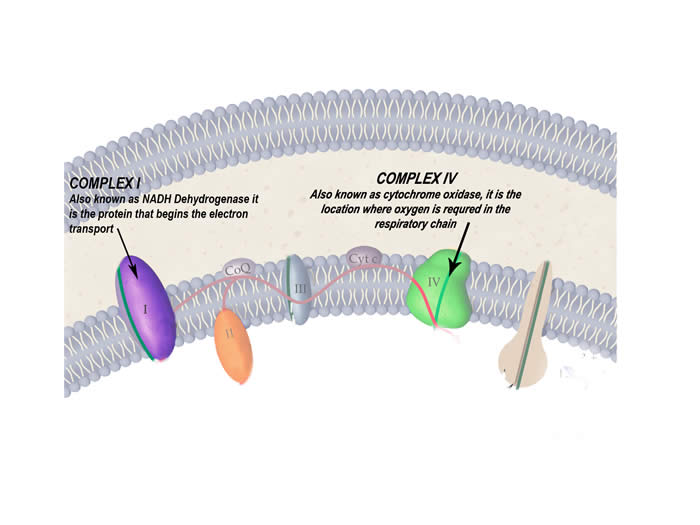
The protein complexes of the respiratory chain that are particularly affected by this disease
- DNA testing from blood reveals a mutation in mtDNA at base pair 3243 for the tRNALeu gene
Is there prenatal testing available?
Prenatal testing for most mitochondrial disorders is available. Molecular testing for prenatal diagnosis is available only when the familial mutation(s) have been found; however, the availability of testing also depends on the mode of inheritance of the condition.
When there is a known genetic diagnosis in the family, it is important for a couple to meet with a genetic counsellor prior to becoming pregnant. This enables the couple to plan in advance, as genetic testing can be a lengthy process.
It is important to note that mitochondrial conditions caused by mutations in mitochondrial DNA (mtDNA) have limited use in prenatal diagnosis, due to principles of heteroplasmy and threshold effect discussed in the inheritance section.
Questions regarding your specific genetic diagnosis and the availability of prenatal diagnosis should always be discussed with a genetic counsellor and/or your obstetrician.
Biological Basis of the Disease
- Genetic changes in the DNA of the mitochondria – most commonly a point mutation at base pair 3243, for the tRNALeu gene
- An A is replaced by a G, therefore an A-T pair becomes a G-C pair
- This changes the tRNA molecule slightly
- This video further explains the point mutation: http://www.youtube.com/watch?v=vNWwSL55gUM&feature=related
- tRNA molecules are responsible for bringing amino acids together to make proteins
- A genetic change in the tRNALeu gene affects the function of these molecules
- The cannot properly make proteins including those of the respiratory chain which creates dysfunctional mitochondria
- Vital organs like the brain and muscles become energy deprived
- Nerve and muscle cells are very energy demanding, and die if they are not provided with ATP
- This results in the many neuromuscular problems that MELAS patients suffer
- Improper functioning of the mitochondria also creates an excess of mitochondrial acids such as pyruvate and lactate to be in body fluids like the blood and CSF, and disrupt the chemical balance in the body
How is it inherited?
The mutation for MELAS occurs in mitochondrial DNA and therefore it follows the pattern of maternal or mitochondrial inheritance.
Treatments
- Although prognosis may be poor, many treatment options are available to improve the daily lifestyle of MELAS patients.
- Medical care under the following specialists may be required: neurologist, medical geneticist, ophthalmologist and cardiologist.
- Nutrition plays a vital role in the management of metabolic disorders and mitochondrial disorders.
- Vitamin and mineral supplements are often prescribed with variable success from patient to patient. The important components of this prescribed “vitamin cocktail” include riboflavin, Coenzyme Q and vitamins E, and C.
- Arginine (an amino acid) supplementation has been found to be helpful in acute stroke like episodes and also for chronic prevention of stroke like episodes.
- Lactic acidosis is treated by sodium bicarbonate or sodium citrate.
- Vitamins help reduce tissue damage caused by the faulty proteins of the mitochondria.
- Seizures can be treated for the most part with traditional anticonvulsant drugs.
- Surgical measures may be required to treat hearing loss.
- Physical therapy and occupational therapy can help improve mobility and comfort
Prognosis
Prognosis for the most part is poor, although it varies greatly from patient to patient. Severity of complications is what eventually determines the life expectancy.
MERRF
What’s in the name?
- MERRF – Myoclonic epilepsy with ragged red fibres
- Myoclonus
- The sudden contraction of muscles, causing brief jerking movements
- Epilepsy
- The condition of having severe, recurrent seizures
- Ragged red fibres Abnormal mitochondria accumulated under the muscle membrane, appear as red strands in muscles, when stained
Who is affected?
MERRF generally affects children and adolescents or even adults at times, and almost always occurs after normal development in infancy. MERRF is very rare with prevalence being less than 1 in 100 000. No ethnic or gender predisposition is found in MERRF.
Symptoms
Neurological:
- Myoclonus – The sudden contraction of muscles, causing brief jerking movements is often the primary symptom
- Generalized Epilepsy – the condition of having frequent, unprovoked seizures
- Ataxia – Gross lack of coordination in muscle movements
- Dementia – a deficit in intellectual ability
Ocular:
- Retinitis Pigmentosa – a group of diseases characterized by
- Swelling of cells in the retina
- Vision loss in the night
- Peripheral vision loss
- Central vision loss
- External ophthalmoplegia/ophthalmoperesis
- Paralysis/weakness of the muscles that open eyelids and move the eyes, resulting in restricted eye movement and droopy eyes
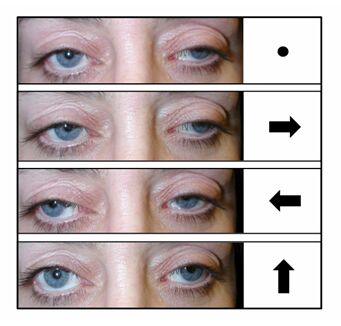
Physical Appearance:
- Short stature (may be present)
Other Symptoms:
- Exercise Intolerance
- Cardiomyopathy - disease affecting the heart muscle, which may lead to heart failure
- Lipomas - benign tumours found beneath the skin, that are composed of fatty tissue
Testing
Laboratory Test:
- Body fluids:
- A higher than normal lactic acid and pyruvic acid (acids found in the mitochondria) concentration in the cerebrospinal fluid (CSF) and blood
- An increase in protein concentration in CSF than normal
- Muscle biopsy:
- Mitochondria from muscle biopsy appear as ragged red fibres
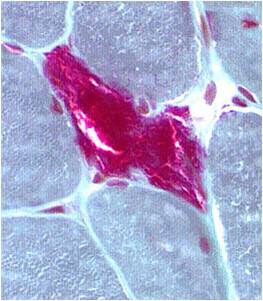
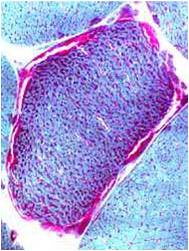
Chemical staining identifies diseased mitochondria, appearing as “ragged red fibres”. Left and right show lower and higher magnification, respectively.
- Brain scans:
- Brain MRI shows basal ganglia calcification (hardening) and atrophy (decrease in size) of certain brain areas
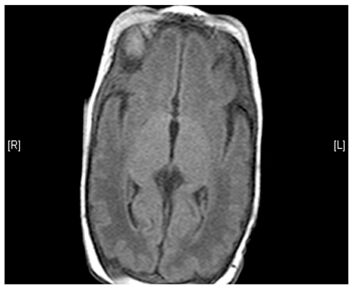
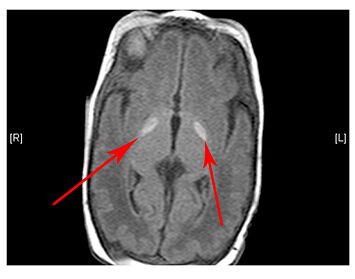
(Left) MRI scan of a healthy brain. (Right) The basal ganglia region of the brain shows abnormal signals.
- ECG tests often show abnormalities in heart function
- Testing enzymes of the respiratory chain often reveal defects in particular proteins – complex I and IV are common in MERRF
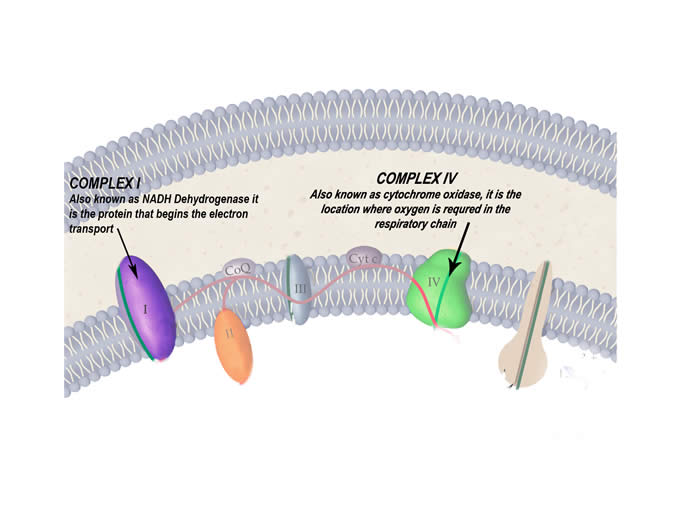
The protein complexes of the respiratory chain that are particularly affected by this disease
- DNA Studies:
- Abnormal mitochondrial mutation at base pair 8344, for tRNALys gene
Is there prenatal testing available?
Prenatal testing for most mitochondrial disorders is available. Molecular testing for prenatal diagnosis is available only when the familial mutation(s) have been found; however, the availability of testing also depends on the mode of inheritance of the condition.
When there is a known genetic diagnosis in the family, it is important for a couple to meet with a genetic counsellor prior to becoming pregnant. This enables the couple to plan in advance, as genetic testing can be a lengthy process.
It is important to note that mitochondrial conditions caused by mutations in mitochondrial DNA (mtDNA) have limited use in prenatal diagnosis, due to principles of heteroplasmy and threshold effect discussed in the inheritance section.
Questions regarding your specific genetic diagnosis and the availability of prenatal diagnosis should always be discussed with a genetic counsellor and/or your obstetrician.
Biological Basis of the Disease
- Mutations in the DNA of the mitochondria is the cause – most commonly a point mutation at base pair 8344, for the tRNALys gene
- An A is replaced by a G, therefore an A-T pair becomes a G-C pair
- This video link explains the point mutation further: http://www.youtube.com/watch?v=vNWwSL55gUM&feature=related
- Molecules of tRNA are responsible for bringing amino acids together to make proteins
- A genetic change in the tRNALeu gene affects the function of these molecules
- The cannot properly make proteins including those of the respiratory chain which creates dysfunctional mitochondria
- Like most mitochondrial diseases, MERRF also disrupts the function of the respiratory chain proteins
- Vital organs like the brain and muscles become energy deprived
- Nerve and muscle cells are very energy demanding, and die if they are not provided with ATP
- This creates the many neuromuscular symptoms that MERRF patients suffer
- Improper functioning of the mitochondria also creates an excess of mitochondrial acids such as pyruvate and lactate to be in body fluids like the blood and CSF, and disrupt the chemical balance in the body
How is it inherited?
The mutation for MERRF occurs in mitochondrial DNA and therefore it follows the pattern of maternal or mitochondrial inheritance.
Treatments
- Medical care under the following specialists may be required: neurologist, medical geneticist, ophthalmologist, cardiologist, and audiologist
- Coenzyme Q10 and L-carnitine are often used to help the dysfunctional proteins of the respiratory chain but most treatments target the manifestations of MERRF rather than the disease itself
- Lactic acidosis is treated by sodium bicarbonate or sodium citrate
- Traditional anticonvulsant drugs are used to treat the epilepsy
- Physical therapy is used to improve motor function
- Mild to moderate aerobic exercise, depending upon tolerance, is recommended for all mitochondrial disease patients
Prognosis
There is extreme variance in the prognosis of those with MERRF. Severity, rate of progression, age of onset, and many other factors determine this.
NARP
What's in the name?
- NARP - Neuropathy, Ataxia, Retinitis Pigmentosa
- Neuropathy – disease of the peripheral nervous system
- Ataxia – without balance or coordination
- Retinitis Pigmentosa
- Retinitis – inflammation of the retina
- Pigmentosa – deposition of pigments on the retina
- This is a group of diseases which can lead to blindness
Who is affected?
Onset of the disease is generally in childhood or early adulthood. NARP is very rare, and its prevalence is actually unknown. There seems to be no ethnic or gender predisposition however.
Symptoms
Neurological Symptoms:
- Developmental delay
- Seizures
- Ataxia – inability to maintain normal posture and smoothness of movement
- Sensory neuropathy – pain, numbness, tingling sensation in the arms and legs
Muscular:
- General muscle weakness and fatigue
- Exercise intolerance
Retinitis Pigmentosa:
- Night blindness
- Tunnel vision
- Loss of central vision caused by damage to the light sensing photoreceptor cells of the retina known as rods and cones (usually damage to the rods)
Testing
Lab Testing:
- Muscle biopsy:
- Abnormal mitochondria from muscle tissue may appear as “ragged red fibres” under a microscope
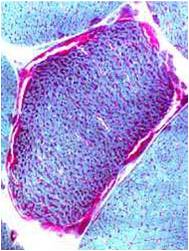
Chemical staining identifies diseased mitochondria, appearing as “ragged red fibres”. Left and right show lower and higher magnification, respectively.
- Body Fluids:
- Elevated concentration of lactic acid in cerebrospinal fluid especially
- Blood plasma shows a higher than normal concentration of the amino acid alanine
- Brain Scans:
- Magnetic resonance spectroscopy (MRS) can detect higher than normal lactic acid concentration in certain brain areas
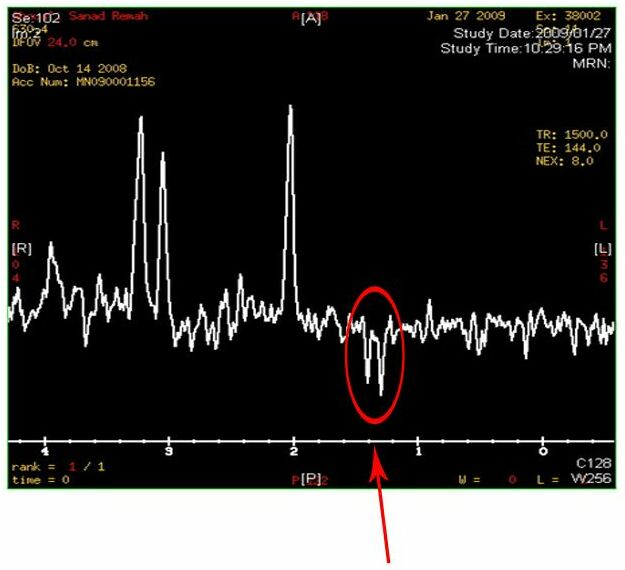
Two inverted peaks that resemble a “W” at around 1.2 on the horizontal axis indicate a high lactate concentration
- Brain Scans continued:
- Atrophy (a decrease in size) of the cerebrum and cerebellum is sometimes seen
- DNA testing in blood usually reveals a T-G or T-C point mutation to the MT-ATP6 gene of the mitochondria
Is there prenatal testing available?
Prenatal testing for most mitochondrial disorders is available. Molecular testing for prenatal diagnosis is available only when the familial mutation(s) have been found; however, the availability of testing also depends on the mode of inheritance of the condition.
When there is a known genetic diagnosis in the family, it is important for a couple to meet with a genetic counsellor prior to becoming pregnant. This enables the couple to plan in advance, as genetic testing can be a lengthy process.
It is important to note that mitochondrial conditions caused by mutations in mitochondrial DNA (mtDNA) have limited use in prenatal diagnosis, due to principles of heteroplasmy and threshold effect discussed in the inheritance section.
Questions regarding your specific genetic diagnosis and the availability of prenatal diagnosis should always be discussed with a genetic counsellor and/or your obstetrician.
Biological basis of the disease
- Caused by a T-G or T-C mutation to the MT-ATP6 gene of the mitochondria
- MT-ATP6 codes part of ATP Synthase, the last component of the respiratory chain
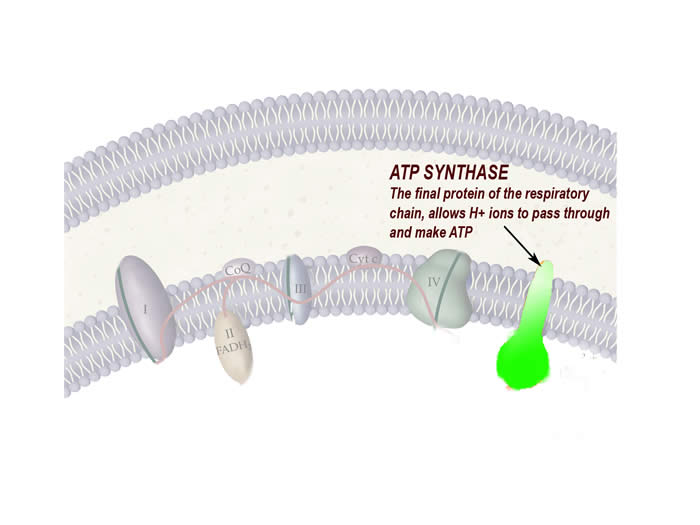
- The consequence is an altered ATP Synthase function, that doesn’t allow ATP production in all cells that contain this diseased mitochondria
- The effects are similar to most mitochondrial point mutation diseases which include neurological symptoms, muscle weakness, and diseases of the eye
How is it inherited?
The mutation for NARP occurs in mitochondrial DNA and therefore it follows the pattern of maternal or mitochondrial inheritance.
Treatments
- High doses of CoQ-10 are prescribed to treat ataxia
- Coenzyme Q-10 is an important component to help the faulty proteins of the respiratory chain function properly, especially complex II.
- Lactic acidosis is treated by sodium bicarbonate or sodium citrate
- To treat retinitis pigmentosa, Vitamin A and E are prescribed
- Antioxidant drugs are used to prevent damage to retinal cells by molecules known as reactive oxygen species (ROS)
Prognosis
There is extreme variance in the prognosis of those with NARP. Severity, rate of progression, age of onset, and many other factors determine this. The condition of patients can be stable for many years, but progression of the disease worsens the symptoms.
LHON
What's in the name?
- LHON – Leber’s Hereditary Optic Neuropathy
- Optic Neuropathy – a disease affecting the optic nerve
- Named after German ophthalmologist Alfred Theodor Leber who first described it in the late 1800s
Who is affected?
The onset of LHON is usually when people are in their late teens or mid twenties. For an unknown reason, males are affected four times as frequently as females. The prevalence of LHON is about 1 in 40000 people. Majority of people (up to 85% in females and 50% in males) with the mutations for LHON, actually do not get LHON, suggesting strong environmental factors.
Symptoms
LHON can result in complete blindness in one or both eyes, within about 6 months of initial symptoms. Along with the common ophthalmological symptoms, other neurological symptoms that are characteristic of mitochondrial disorders are also present.
Ophthalmological Symptoms:
- Pain, discomfort, tingling sensation in and around both eyes
- Visual field defects, which decreases peripheral vision
- Decrease in sharpness in sight, called visual acuity
- Decrease in colour vision
- Progressive loss of central vision, eventually resulting in complete blindness
Other Symptoms:
- Developing neurological features similar to multiple sclerosis
- Movement disorders such as tremors, muscle contractions, and twisted repetitive movements known as dystonia
- Poor coordination
- Numbness in limbs
- Muscle weakness
- Cardiac conduction block - Abnormalities in the electrical impulses that control heartbeat
Testing
Lab testing:
- Geneticists can identify the exact change in DNA from the blood, and the diagnosis is almost 100% accurate, once blindness has already occurred
- Testing enzymes of the respiratory chain often reveal defects in complex I
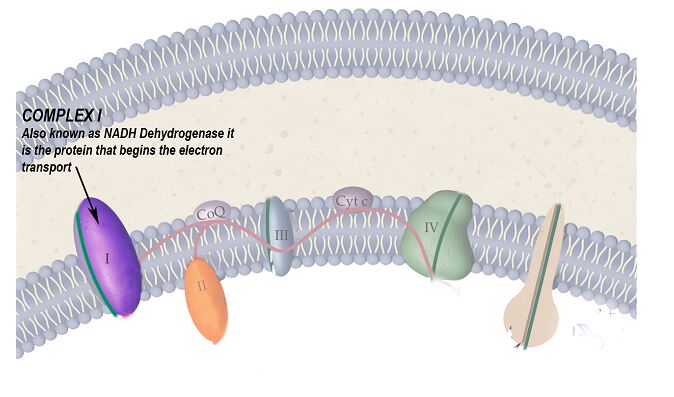
The protein complex of the respiratory chain that is particularly affected by this disease
Is there prenatal testing available?
Prenatal testing for most mitochondrial disorders is available. Molecular testing for prenatal diagnosis is available only when the familial mutation(s) have been found; however, the availability of testing also depends on the mode of inheritance of the condition.
When there is a known genetic diagnosis in the family, it is important for a couple to meet with a genetic counsellor prior to becoming pregnant. This enables the couple to plan in advance, as genetic testing can be a lengthy process.
It is important to note that mitochondrial conditions caused by mutations in mitochondrial DNA (mtDNA) have limited use in prenatal diagnosis, due to principles of heteroplasmy and threshold effect discussed in the inheritance section.
Questions regarding your specific genetic diagnosis and the availability of prenatal diagnosis should always be discussed with a genetic counsellor and/or your obstetrician.
How is it inherited?
The mutation for LHON occurs in mitochondrial DNA and therefore it follows the pattern of maternal or mitochondrial inheritance.
Biological basis of the disease
- Caused by one of the three following point mutations in the mitochondrial DNA:
- G-A at nucleotide 3460
- G-A at nucleotide 11778
- T-C at nucleotide 14484
- These genetic changes may affect more than one protein of the respiratory chain thereby decreasing the effectiveness of the mitochondria to produce ATP
- Complex I is the protein commonly affected by this mutation
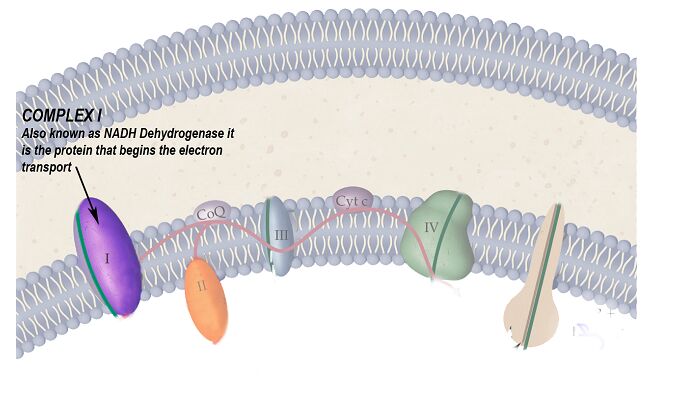
The protein complex of the respiratory chain that is particularly affected by this disease
- These mutations also increase the risk of making molecules known as reactive oxygen species (ROS) which damage mtDNA
- Optic nerve cells are unable to produce energy, and cannot function, resulting in cell death causing blindness
Treatments
- No current cure or effective treatments are available to reverse or improve vision loss from LHON
- Experimental trails with vitamin B12 and C supplementation and quinone are currently being tested, with limited success
- Those with LHON in their families, are of course at a higher risk for developing it themselves, and are advised to avoid smoking and heavy alcohol consumption which may act as environmental triggers
Prognosis
There is extreme variance in the prognosis of those with LHON. Severity, rate of progression, age of onset, and type of mutation are just a few of the factors that determine this, however blindness is almost always certain. The mutation “T>C 14484” is generally associated with the best prognosis.
Leigh's Disease
What’s in the name?
- Leigh’s Disease is named after the neurologist who first described it in 1951
- Also known as Subacute Necrotizing Encephalomyelopathy (SNEM)
- Subacute - a rather sudden onset of a disease
- Necrotizing - causing death to a certain area or tissue
- Encephalo – -pertaining to the brain and nervous system
- –myelopathy – disease of the spine, causing dysfunction
- Leigh’s disease therefore damages tissue of the nervous system causing neurological problems, particularly with motor control
Who is affected?
The onset of the disease is usually in the first year of life. Later onset (after infancy) generally means a slower progression of the disease. The prevalence of Leigh’s disease is about 1 in 35 000. There is no ethnic or gender predisposition to the disease.
Symptoms
The brain and muscles are most affected which is very typical of mitochondrial disease.
Neurological:
- Lack of motor control (cannot hold toys in hands etc)
- Dementia – loss of intellectual ability
- Seizures
Musculoskeletal:
- Generalized weakness
- Exercise intolerance
- Lack of muscle tone
Other:
- Loss of appetite
- Vomiting
- Overall irritability and continuous crying
- Kidney and respiratory failure
Testing
Lab Testing:
- Muscle biopsy:
- Mitochondria appear as ragged red fibres under a microscope
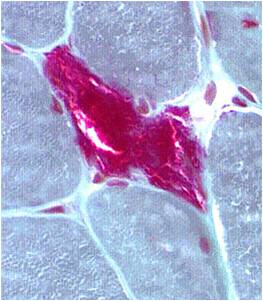
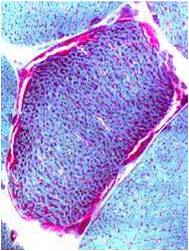
Chemical staining identifies diseased mitochondria, appearing as “ragged red fibres”. Left and right show lower and higher magnification, respectively.
- Body fluids:
- Elevated concentration of lactic acid in cerebrospinal fluid especially
- Blood Plasma shows a higher than normal concentration of the amino acid alanine
- Brain scans:
- Chemical staining identifies diseased mitochondria, appearing as “ragged red fibres”. Left and right show lower and higher magnification, respectively.
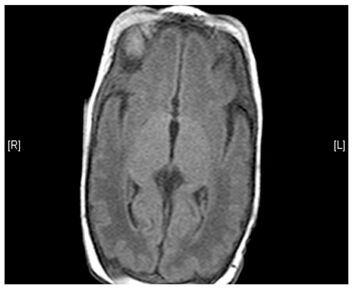
(Left) MRI scan of a healthy brain. (Right) The basal ganglia region of the brain shows abnormal signals.
- Testing enzymes of the respiratory chain often reveal defects in particular proteins – complex I, IV are common
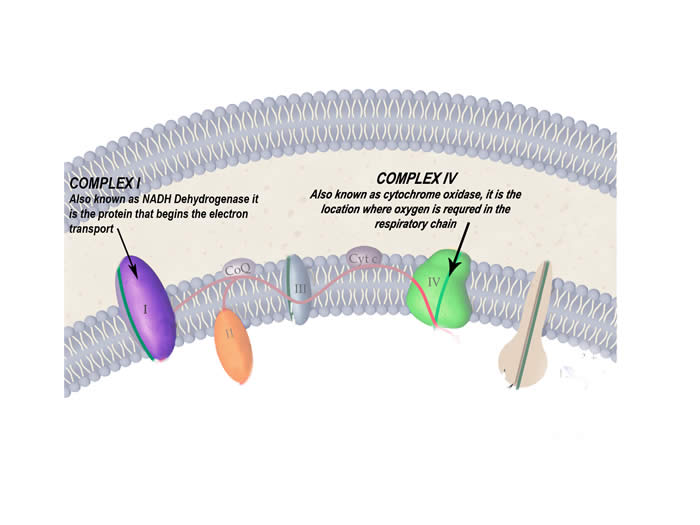
The protein complexes of the respiratory chain that are particularly affected by this disease
- DNA testing shows one of many possible mutations in mitochondrial DNA genes – a common mutation is T8993G or T8993C MT-ATP6
Is there prenatal testing available?
Prenatal testing for most mitochondrial disorders is available. Molecular testing for prenatal diagnosis is available only when the familial mutation(s) have been found; however, the availability of testing also depends on the mode of inheritance of the condition.
When there is a known genetic diagnosis in the family, it is important for a couple to meet with a genetic counsellor prior to becoming pregnant. This enables the couple to plan in advance, as genetic testing can be a lengthy process.
It is important to note that mitochondrial conditions caused by mutations in mitochondrial DNA (mtDNA) have limited use in prenatal diagnosis, due to principles of heteroplasmy and threshold effect discussed in the inheritance section.
Questions regarding your specific genetic diagnosis and the availability of prenatal diagnosis should always be discussed with a genetic counsellor and/or your obstetrician.
Biological basis of the disease and the mechanism of inheritance
There are many ways that Leigh’s disease can occur, each with a distinct genetic change.
X Linked:
- A genetic change is carried on the X chromosome
- Most individuals with Leigh’s disease do not reproduce:
- Therefore only carriers can pass down the mutation in Leigh’s disease and since only females can be carriers in X-linked diseases, only females can pass down the Leigh’s disease mutation
- This particular genetic change causes defect in the pyruvate dehydrogenase enzyme, which is required for food molecules (particularly glucose from carbohydrates) to enter the mitochondria
Autosomal Recessive:
- Both parents must be carriers of the disease in order to pass it down to the next generation
- This mutation affects the gene for the protein called cytochrome-c-oxidase (COX), the fourth protein in the respiratory chain
Mitochondrial:
The final method for inheritance of Leigh’s disease is maternally, due to one of many point mutations in mitochondrial DNA. This follows a maternal, or mitochondrial pattern of inheritance
All methods of inheritance of Leigh’s disease present the same end result (i.e. dysfunctional mitochondria, and decreased ATP production). This results in energy deficits under stressful conditions within neurons, in particular regions of the nervous system, and leads to the symptoms described above.
Treatments
- Consultation with a metabolic geneticist, neurologist, and cardiologist may be required
- Most treatments target controlling the symptoms rather than the disease itself
- Lactic acidosis is treated by sodium bicarbonate or sodium citrate
- Traditional anticonvulsant drugs are used to treat seizures
- A “vitamin cocktail” consisting of riboflavin, thiamine, and coenzyme Q10, and vitamin B1 is used to improve mitochondrial function
- A high fat diet is often recommended due to the enzyme pyruvate dehydrogenase deficiency
Prognosis
Unfortunately, prognosis is generally poor although it largely depends on the type of mutation. Pyruvate dehydrogenase and complex IV deficiency is associated with the worst prognosis. Those with less severe forms of the disease may have a longer lifespan.
Kearns-Sayre
What’s in the name?
Kearns-Sayre syndrome is also referred to as Chronic Progressive External Ophthalmoplegia and Myopathy or Ophthalmoplegia, Pigmentary Degeneration of the Retina and Cadiomyopathy
- Ophthalmoplegia – the paralysis of ocular muscles
- Myopathy – a disease of muscle tissue
- Cardiomyopathy – a disease of the cardiac muscle
Hence, Kearns-Sayre syndrome affects various energy demanding muscles and nerves in the body. Heart muscle and the many muscles controlling eye movement are particularly affected.
Who is affected?
The disease usually occurs in early childhood, and almost always before 20 years of age. Kearns-Sayre disease is extremely rare with prevalence being about 1-3 in 100 000 individuals. No ethnic or gender predisposition exists for Kearns-Sayre disease.
Symptoms
Kearns-Sayre syndrome has symptoms characteristic of most other mitochondrial disease however the eyes are especially affected.
Ocular:
- Chronic progressive external ophthalmoplegia (CPEO) – progressive paralysis of muscles in and around the eyes causing:
- restricted movement of the eyes
- the appearance of drooping eyelids
- Retinitis pigmentosa – accumulation of coloured pigments on the retina, at the back of the eye, which can lead to blindness
- Inflammation of the nerves on the retina
- Degeneration of retinal tissue
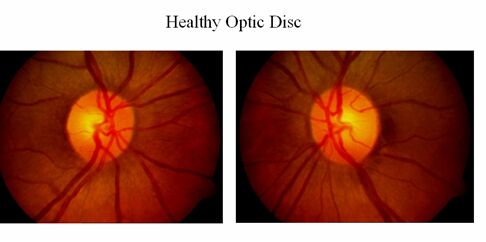
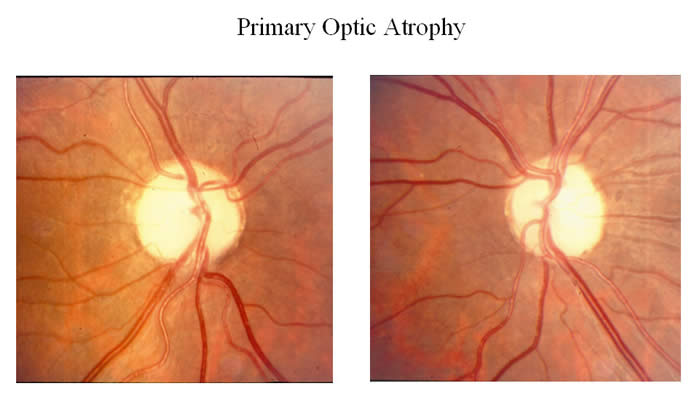
Other Symptoms:
- Short, stooped stature
- Cardiac conduction block - problems in the electrical system of the heart
- Ataxia - uncoordinated movements in the limbs as well as loss of balance
- Dementia - loss of intellectual ability
- Hearing loss
- Exercise intolerance
- Kidney dysfunction
Endocrine symptoms:
- Diabetes mellitus
- Hypoparathyroidism
- Growth hormone deficiency
Testing
Lab Testing:
- Body fluids:
- Elevated levels of lactic acid and pyruvic acid (mitochondrial acids) in the blood and cerebrospinal fluid (CSF)
- Muscle biopsy:
- Testing using electromyogram (EMG) indicate muscle disease
- Mitochondria from muscle tissue appear as “ragged red fibres” under a microscope
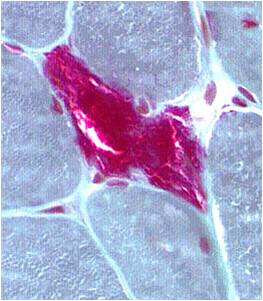
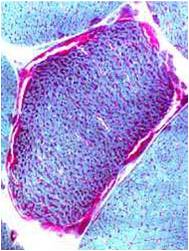
Chemical staining identifies diseased mitochondria, appearing as “ragged red fibres”. Left and right show lower and higher magnification, respectively.
- Brain scans:
- Atrophy in various regions of the brain
- MRI scans may show destruction of an area of the brain called white matter, a condition called leukoencephalopathy
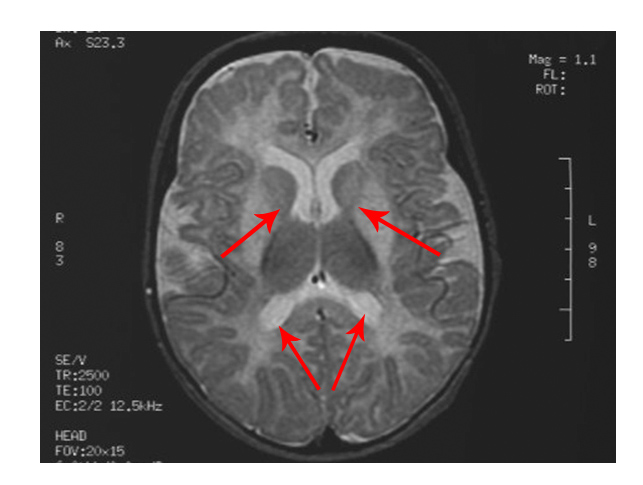
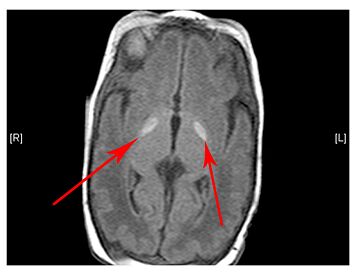
The scan shows changes in white matter within the brain as well as symmetrical abnormalities in signals (white areas) in the area known as the putamen (bottom arrows), in both hemispheres
- Cardiac:
- Electrocardiogram (ECG) is used to test heart function
- Testing enzymes of the respiratory chain often reveal defects in particular proteins – complex IV is especially affected in Kearns-Sayre
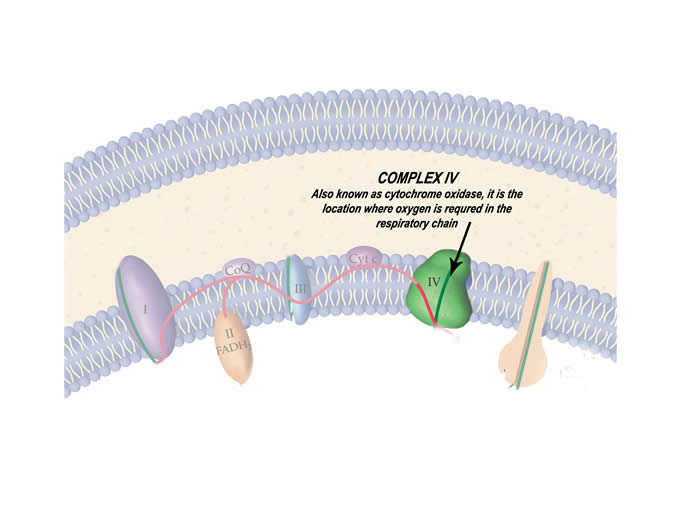
The protein complex of the respiratory chain that is particularly affected by this disease
- DNA testing reveals mitochondria that have deletions in large amounts to their DNA
Is there prenatal testing available?
Prenatal testing for most mitochondrial disorders is available. Molecular testing for prenatal diagnosis is available only when the familial mutation(s) have been found; however, the availability of testing also depends on the mode of inheritance of the condition.
When there is a known genetic diagnosis in the family, it is important for a couple to meet with a genetic counsellor prior to becoming pregnant. This enables the couple to plan in advance, as genetic testing can be a lengthy process.
It is important to note that mitochondrial conditions caused by mutations in mitochondrial DNA (mtDNA) have limited use in prenatal diagnosis, due to principles of heteroplasmy and threshold effect discussed in the inheritance section.
Questions regarding your specific genetic diagnosis and the availability of prenatal diagnosis should always be discussed with a genetic counsellor and/or your obstetrician.
How is it inherited?
- Kearns-Sayre disease, when inherited, follows a maternal or mitochondrial pattern of inheritance
- Interestingly however, Kearns-Sayre disease unlike most other mitochondrial disorders is rarely inherited (1 in 24 chance a baby will be affected if the mother has the disease) – most often it occurs spontaneously in a sporadic mutation
Biological basis of the disease
- Large scale deletions of mitochondrial DNA, at varying areas of the genome
- A common deletion is of approximately 1/3 of the genome
- In mtDNA deletions, many essential mitochondrial genes are lost, particularly those of the respiratory chain
- This affects mitochondrial function, and energy production
- Vital organs like the brain and muscles become energy deprived
- Nerve and muscle cells are very energy demanding, and die if they are not provided with ATP
This results in the many neuromuscular problems that Kearns-Sayre patients suffer
Treatments
- No cure is available to reverse or slow the progression of the disease, but treatments are available to control the symptoms
- Consultation with an ophthalmologist, neurologist, endocrinologist and cardiologist are usually required
- Cardiac pacemaker to treat heart conduction block
- Surgery may be necessary for severe drooping eyelids
- Lactic acidosis is treated by sodium bicarbonate or sodium citrate
- Administration of coenzyme Q10, L-carnitine and antioxidants to treat defective proteins of the respiratory chain
- Hormone replacement therapy for disorders of the endocrine system
- Physical therapy and occupational therapy to improve mobility
- Mild to moderate physical activity is also recommended for patients with muscle weakness
Prognosis
Prognosis varies widely from patient to patient due to severity, amount of damage to organs and other factors. Kearns-Sayre is a degenerative disease with slow progression. Unfortunately most patients have a life expectancy much shorter than average.
MNGIE
What's in the name?
- MNGIE – Mitochondrial neurogastrointestinal encephalopathy
- Neurogastrointesintal – the brain and organs of the digestive system are affected
- Encephalo-pathy – disease of the nervous system
- Alternate names for MNGIE are
- Myoneurogastrointestinal encephalopathy syndrome
- Oculogastrointestinal muscular dystrophy
Who is affected?
MNGIE is extremely rare with only about 70 cases reported worldwide. It usually affects children and adolescents before the age of 20. There is no ethnic or gender predisposition for MNGIE.
Symptoms
MNGIE presents a wide spectrum of different symptoms of the nervous, gastrointestinal and muscular systems. The symptoms may begin presenting themselves at any age however children and adolescents under 20 are most commonly affected.
Gastrointestinal:
- Dysmotility – Muscles of the gastrointestinal tract do not function effectively due to problems in the nerves controlling the muscles. This causes:
- Feeling full after only eating small amounts
- Dysphagia – difficulty swallowing
- Nausea and vomiting
- Abdominal pain
- Diarrhea
Neurological:
- Peripheral neuropathy – tingling, numbness and weakness in the limbs
- External ophthalmoplegia/ophthalmoperesis – paralysis of muscles in and around the eyes causing restricted eye movement and drooping eyelids
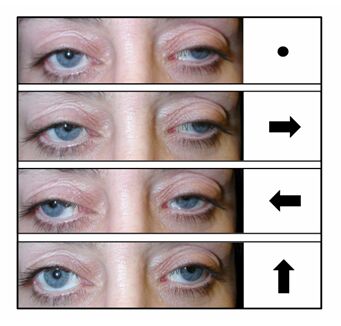
- Leukoencephalopathy – a disease causing destruction of an area of the brain called white matter
Appearance:
- Significant weight loss
- Reduced muscle mass
- Short stature
Testing
Lab Testing
- Body fluids
- A change in particular enzyme concentration or activity in the blood:
- An increase in plasma thymidine concentration
- An increase in deoxyuridine concentration
- Decreased activity of thymidine phosphorylase
- Lactic Acidosis - an increased concentration of lactic acid in blood and cerebrospinal fluid
- The following link provides detailed information and videos on obtaining CSF via lumbar puncture: http://emedicine.medscape.com/article/80773-treatment
- A change in particular enzyme concentration or activity in the blood:
- Muscle biopsy
- After chemical staining, diseased mitochondria appear as ragged red fibres under a microscope
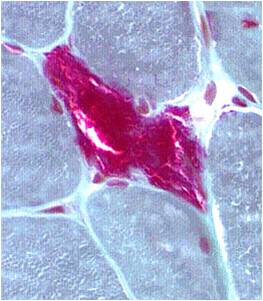
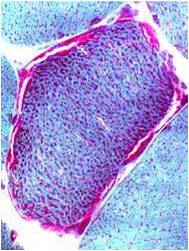
Chemical staining identifies diseased mitochondria, appearing as “ragged red fibres”. Left and right show lower and higher magnification, respectively.
- Brain scans
- MRI scans reveal a destruction in an area of the brain known as white matter, a condition called leukoencephalopathy
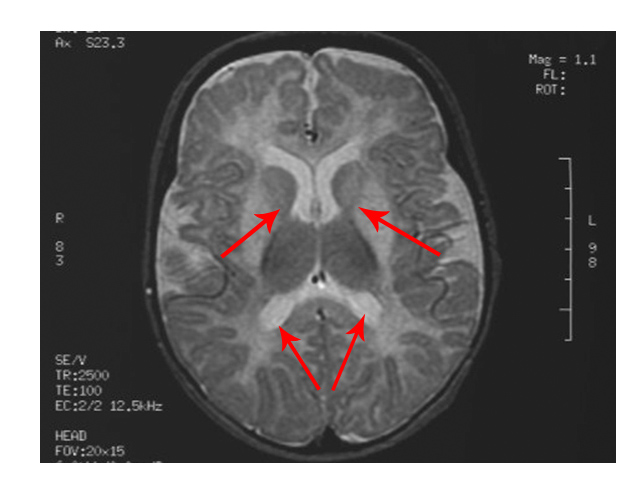
The scan shows changes in white matter within the brain as well as symmetrical abnormalities in signals (white areas) in the area known as the putamen (bottom arrows), in both hemispheres
- EMG tests for nerve conduction studies
- Testing enzymes of the respiratory chain often reveal defects in multiple proteins – complex IV is particularly affected
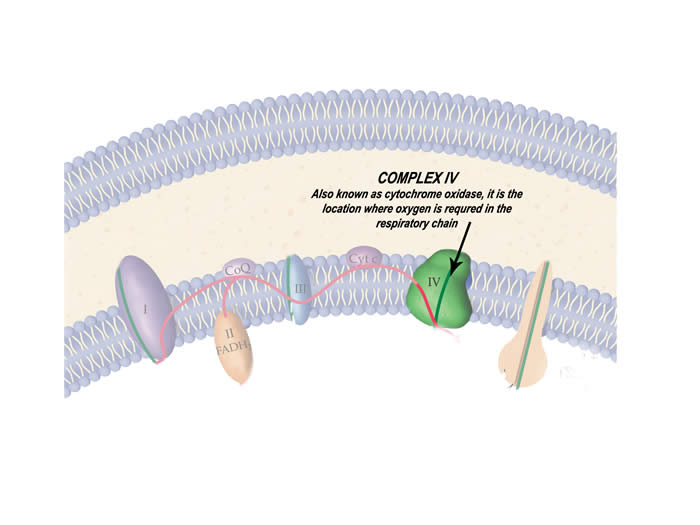
The protein complex of the respiratory chain that is particularly affected by this disease
- DNA testing in blood for TYMP gene, reveals a mutation in nearly 100% of those affected
Is there prenatal testing available?
Prenatal testing for most mitochondrial disorders is available. Molecular testing for prenatal diagnosis is available only when the familial mutation(s) have been found; however, the availability of testing also depends on the mode of inheritance of the condition.
When there is a known genetic diagnosis in the family, it is important for a couple to meet with a genetic counsellor prior to becoming pregnant. This enables the couple to plan in advance, as genetic testing can be a lengthy process.
Questions regarding your specific genetic diagnosis and the availability of prenatal diagnosis should always be discussed with a genetic counsellor and/or your obstetrician.
How is it inherited?
MNGIE follows the pattern of an autosomal recessive form of inheritance.
Biological basis of the disease
- A mutation to a gene in the nucleus called TYMP has been associated with causing MNGIE
- The gene codes for a protein called thymidine phosphorylase, which is essential for regulating the level of the molecule thymidine
- When regulated, small amounts of thymidine are essential for normal mitochondrial function, but a drastic increase in thymidine causes problems to mitochondrial DNA
- Lab tests for MNGIE patients show a decrease in thymidine phosphorylase protein and therefore an increase in thymidine since there is nothing to regulate its levels
- Problems to mitochondrial DNA include:
- Deletions of parts of the DNA
- Duplications to parts of the DNA
- These deletions and duplications affect genes which code for proteins of the respiratory chain
- This creates mitochondria that don’t function properly and as a result muscle and nerve cells become energy deprived
- The exact causes of specific symptoms of MNGIE however are still unknown
Treatments
- Medical care under the following specialists may be required: neurologist, gastroenterologist, medical geneticist, psychiatrist, ophthalmologist
- There is no medication to slow the progression of the disease and treatments only target at controlling the symptoms
- Lactic acidosis is treated by sodium bicarbonate or sodium citrate
- Antiemetic drugs are used to treat nausea and vomiting
- Those with extreme dysphagia may require nutritional support using food tubes
- Dysmotility may cause intestinal bacterial growth, and antibiotics may be required
- Physical therapy and occupational therapy help increase mobility and comfort
- To treat the excess thymidine levels, stem cell (bone marrow) transplants have been used
Prognosis
Prognosis is generally quite poor. As the disease progresses, symptoms gradually worsen. Unfortunately, MNGIE patients’ lifespan is much shorter than average.
Pearson's Syndrome
What’s in the name?
- Pearson’s syndrome was named after the physician who first described it in 1979
- Alternate names for Pearson’s syndrome are Pearson’s marrow pancreas syndrome or Sideroblastic anemia with marrow cell vacuolization and exocrine pancreatic dysfunction
- Sideroblast - a nucleated red blood cell which contains excess iron, and is found in the bone marrow
- Anemia - a low count in haemoglobin protein, which is used to carry iron
- The body has plenty of iron available yet cannot use it due to haemoglobin deficiency
- Marrow cell vacuolization – the formation of vacuoles (membrane bound structures inside cells that store many chemicals) in bone marrow cells
- Exocrine pancreas – the component of the pancreas that releases enzymes via ducts in the intestines, during digestion
Who is affected?
The onset of Pearson’s is most common during infancy or early childhood. It is extremely rare with only about 80 cases reported worldwide. There is no ethnic or gender predisposition to the disease.
Symptoms
Neuromuscular:
- Tremor
- Lack of muscle tone
- Fatigue
Digestive:
- Chronic diarrhoea
- Vomiting episodes
- Fatty stool
- Liver failure
Other symptoms (may be present):
- Pallor - pale skin which is generally caused by anemia
- Erythema – redness of the skin
- Sensitivity to light
- Short stature
- Weight loss
Testing
Lab Testing:
- Body fluids:
- Abnormally low count of reticulocytes (red blood cells which still have not fully developed), as well as white blood cells and platelets
- Lactic acidosis - Increase in lactic acid concentration (a mitochondrial acid) in the blood and CSF
- Increased glucose, amino acids and other chemicals in the urine which are normally filtered out by kidneys (therefore indicates a kidney problem)
- Biopsy:
- Bone marrow biopsy shows a change in the appearance of bone cells
- Brain scans using MRI and MRS may be ordered for individuals with severe neurological symptoms
- Enzymology:
- Tests on particular liver enzymes such as transaminase
- Pancreatic enzyme tests may be performed on isoamylase, trypsinogen, and lipase enzymes
- DNA testing on white blood cells reveals deletions of mitochondrial DNA
Biological basis of the disease
- Large scale deletions and duplications of mitochondrial DNA
- In mtDNA deletions, many essential mitochondrial genes are lost, particularly those of the respiratory chain proteins
- This affects mitochondrial function, and energy production in cells
- Cells of vital organs, especially of the digestive system become energy deprived and cannot function
How is it inherited?
- Pearson’s disease is caused by mtDNA mutations, which can only be inherited maternally. However, Pearson’s actually more commonly occurs spontaneously due to a sporadic mutation
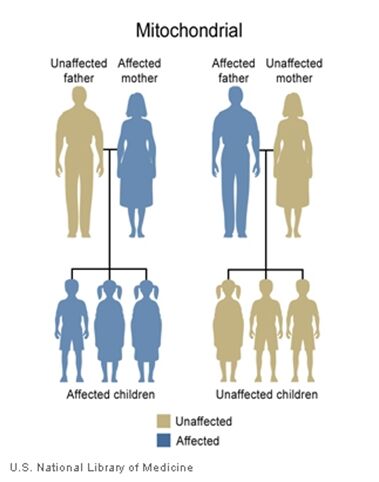
Treatments
- Generally, consultation with a metabolic geneticist and haematologist are required
- There are no treatments for the disease itself, but only to control the symptoms
Sodium bicarbonate is used to treat acidosis - Red blood cell transfusions may be required for the various anemias that may occur as a result of Pearson’s
- Stem cell transplantation has been used effectively in one case of Pearson’s syndrome
- Physical therapy and occupational therapy may be used to improve mobility and comfort
Prognosis
Prognosis may be poor for patients but it actually depends on severity of complications arising from Pearson’s. Life expectancy may also be greatly reduced. Other complications such as symptoms of Kearns-Sayre syndrome are sometimes found in the later stages of Pearson’s.
CPEO
What’s in the name?
- CPEO - Chronic Progressive External Ophthalmoplegia
- Chronic – a medical condition that has developed over an extended period of time, or is long lasting
- Progressive – Symptoms worsen as time passes
- External Ophthalmoplegia – Paralysis of the muscles surrounding the eyes, restricting movement of the eyes and causing the appearance of drooping eyelids
Who is affected?
The disease usually occurs in early childhood, and almost always before 20 years of age. CPEO is extremely rare with prevalence being about 1-3 in 100 000 individuals. No ethnic or gender predisposition exists for CPEO
Symptoms
Ocular:
- External ophthalmoplegia–Paralysis of muscles in and around the eyes causing:
- restricted movement of the eyes
- the appearance of drooping eyelids
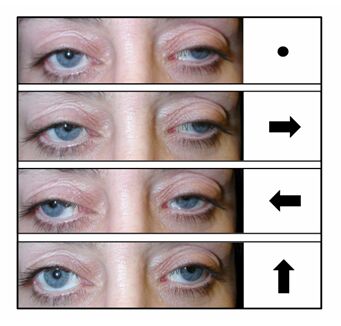
Other Symptoms:
- Short, stooped stature
- Severe weakness in limbs
Often, other symptoms which are characteristic of Kearns-Sayre syndrome (KSS) are present but not enough to be clinically considered KSS. In these cases, CPEO is often referred to as “KSS minus”.
Testing
Lab Testing:
- Body fluids:
- Elevated levels of lactic acid and pyruvic acid (mitochondrial acids) in the blood and cerebrospinal fluid (CSF)
- The following link provides detailed information and videos lumbar puncture, the procedure of obtaining cerebrospinal fluid http://emedicine.medscape.com/article/80773-treatment
- Muscle biopsy:
- Testing using electromyogram (EMG) indicate muscle disease
- Mitochondria from muscle tissue appear as “ragged red fibres” under a microscope
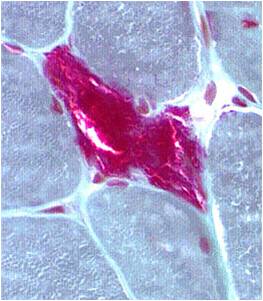
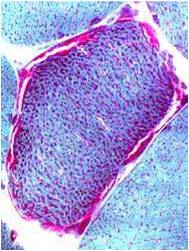
Chemical staining identifies diseased mitochondria, appearing as “ragged red fibres”. Left and right show lower and higher magnification, respectively.
- Brain scans:
- Atrophy in various regions of the brain
- MRI scans may show destruction of an area of the brain called white matter, a condition called leukoencephalopathy
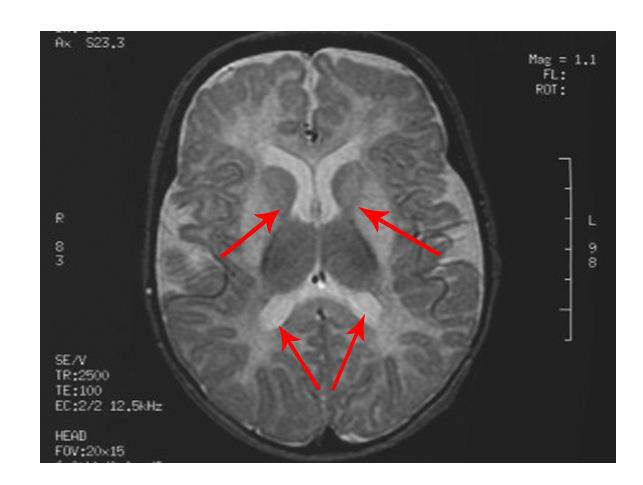
The scan shows changes in white matter within the brain as well as symmetrical abnormalities in signals (white areas) in the area known as the putamen (bottom arrows), in both hemispheres
- Testing enzymes of the respiratory chain often reveal defects in particular proteins – complex IV is especially affected in CPEO
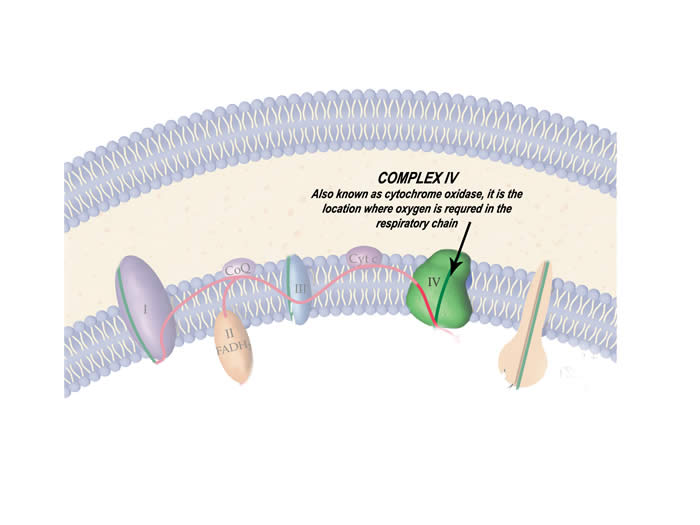
The protein complex of the respiratory chain that is particularly affected by this disease
- DNA testing reveals mitochondria that have deletions in large amounts to their DNA
Is there prenatal testing available?
Prenatal testing for most mitochondrial disorders is available. Molecular testing for prenatal diagnosis is available only when the familial mutation(s) have been found; however, the availability of testing also depends on the mode of inheritance of the condition.
When there is a known genetic diagnosis in the family, it is important for a couple to meet with a genetic counsellor prior to becoming pregnant. This enables the couple to plan in advance, as genetic testing can be a lengthy process.
It is important to note that mitochondrial conditions caused by mutations in mitochondrial DNA (mtDNA) have limited use in prenatal diagnosis, due to principles of heteroplasmy and threshold effect discussed in the inheritance section.
Questions regarding your specific genetic diagnosis and the availability of prenatal diagnosis should always be discussed with a genetic counsellor and/or your obstetrician.
How is it inherited?
Since mitochondrial DNA is affected in CPEO, it follows a mitochondrial or maternal pattern of inheritance
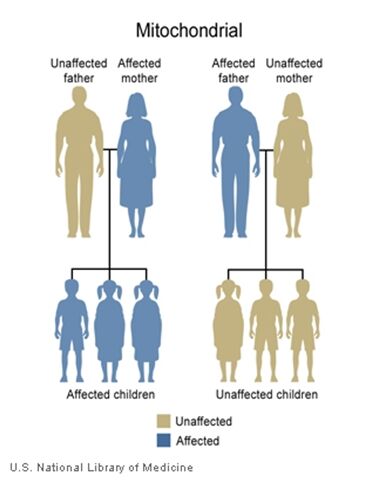
Biological basis of the disease
- Large scale deletions of mitochondrial DNA, at varying areas of the genome
- A common deletion is of approximately 1/3 of the genome
In mtDNA deletions, many essential mitochondrial genes are lost, particularly those of the respiratory chain - This affects mitochondrial function, and energy production
- Energy demanding parts of the body like the eyes and nerves are particularly affected
Treatments
- No cure is available to reverse or slow the progression of the disease, but treatments are available to control the symptoms
- Consultation with a neurologist, ophthalmologist and endocrinologist may be required
- Surgery may be necessary for severe drooping eyelids
- Lactic acidosis is treated by sodium bicarbonate or sodium citrate
- Administration of coenzyme Q10, L-carnitine and antioxidants to treat defective proteins of the respiratory chain
Prognosis
Prognosis varies widely from patient to patient due to severity, amount of damage to organs and other factors. CPEO is a degenerative disease with slow progression. Unfortunately most patients have a life expectancy much shorter than average.
Alpers' Syndrome
What’s in the name?
- Alpers’ disease was named after the physician who first described it in 1931
- Also termed Progressive Neuronal Degeneration of Childhood with Liver Disease and poliodystrophy
- Poliodystrophy – disease of the nervous system caused by a decrease in the area the brain known as grey matter
Who is affected?
Alpers’ disease generally affects young children before the age of five although there have been cases where symptoms begin in adolescence. This is termed as Juvenile Alpers’ disease. The prevalence of Alpers’ is only about 1 in every 200 000. There is no ethnic or gender predisposition to the disease.
Symptoms
Neurological:
- Severe epilepsy which causes the seizures, and is generally the first symptom
- Dementia - a loss of intellectual ability
- A gradual loss of previously learnt skills
- Involuntary movements of the limbs, face and head
- Spastic quadriplegia - inability to use and control movements of the arms and legs, in the later stages of the disease
- Optic atrophy – the degeneration of the optic nerve causing blindness
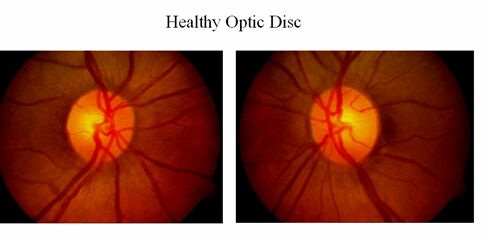
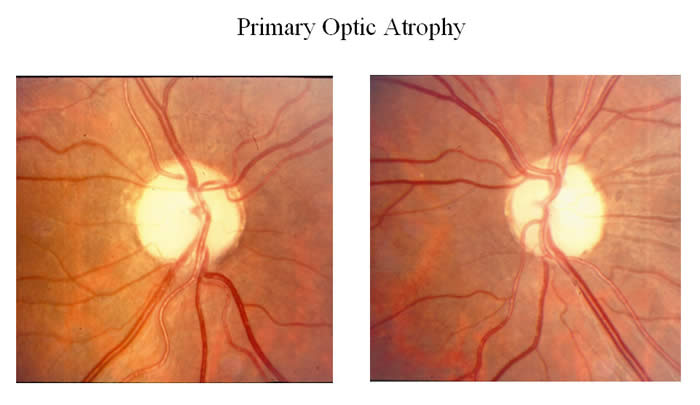
Liver:
- Jaundice - a liver disease causing yellowish discolouration of the skin and eyes
- In rare cases, complete liver failure
Biopsy:
- Liver biopsy test may be helpful to diagnose in later stages of the disease
- EEG tests reveal distinct brain wave patterns
- Confirmed diagnosis of the disease is only available from autopsy of the brain after death
- Autopsy reveals degradation of the area of the brain known as grey matter
Is there prenatal testing available?
Prenatal testing for most mitochondrial disorders is available. Molecular testing for prenatal diagnosis is available only when the familial mutation(s) have been found; however, the availability of testing also depends on the mode of inheritance of the condition.
When there is a known genetic diagnosis in the family, it is important for a couple to meet with a genetic counsellor prior to becoming pregnant. This enables the couple to plan in advance, as genetic testing can be a lengthy process.
Questions regarding your specific genetic diagnosis and the availability of prenatal diagnosis should always be discussed with a genetic counsellor and/or your obstetrician.
How is it inherited?
- Alpers’ disease follows an autosomal recessive pattern of inheritance
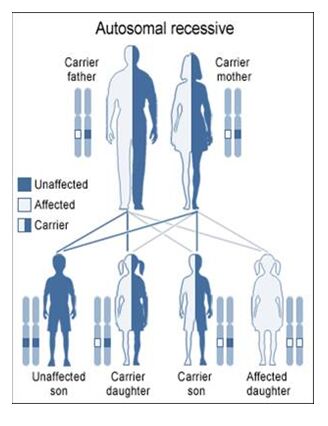
Biological basis of the disease
- A mutation to the gene known as polymerase gamma which makes a protein involved in replication and repair of DNA
- This gene is found in nuclear DNA
- A mutation to this gene causes large scale deletions of mitochondrial DNA
In mtDNA deletions, many essential mitochondrial genes are lost, particularly those of the respiratory chain - This affects mitochondrial function, and therefore energy production
- Energy demanding cells of the body such as those located in the brain or liver often suffer, causing the symptoms seen in Alpers’ disease
Treatments
- Consultation with a neurologist, metabolic geneticist and a hepatologist (liver specialist) may be required
- There is no current cure or treatment to slow the progression of the disease but drugs are prescribed to control the symptoms
- Anticonvulsant medication to treat seizures
- Physical therapy and occupational therapy is used to improve mobility and comfort
Prognosis
There is extreme variance in the prognosis of those with Alpers’ disease. Severity, rate of progression, age of onset, and many other factors determine this, although life expectancy is generally drastically reduced.
Mitochondrial DNA Depletion Syndromes
What’s in the name?
- A single mitochondrion may contain multiple copies of its DNA
- Like its name sake, DNA depletion syndromes causes a reduction in the number of copies of DNA within the mitochondrion
Who is affected?
The onset of the disease is early, with symptoms beginning in infancy (before about 5 months of age). The disease is extremely rare with only about 17 cases reported worldwide. Although various ethnic groups have been affected, a common mutation has been found in Faroese people (an ethnic group with a population of about 25 000 living Scandinavia)
Symptoms
Neurological:
- Psychomotor delay – a delayed development of intellect and motor ability
- Dystonia – uncontrollable muscle contractions causing repetitive twisting movements
- Epilepsy - The condition of having severe, recurrent seizures
- External ophthalmoplegia
- Paralysis of the muscles that open eyelids, and control eye movement
- Results in drooping of the eyelids
Muscular:
- Muscle hypotonia – low muscle tone, and an overall reduced muscle mass
- Results in a lack of control of head and neck movement
Physical Appearance:
- Kyphosis - the appearance of a hunchback
- Scoliosis - a spine that curves side to side in a person
Testing
Lab Testing:
- Body Fluids:
- Elevated levels of lactic acid found in the blood and cerebrospinal fluid
- Brain Scans:
- Atrophy of certain areas of the brain
- Abnormal signals from basal ganglia region of the brain on an MRI
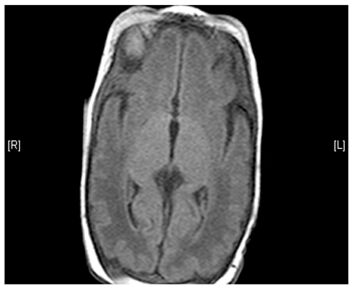
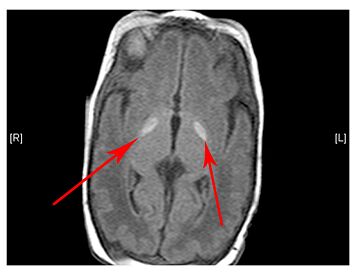
(Left) MRI scan of a healthy brain. (Right) The basal ganglia region of the brain shows abnormal signals.
How is it inherited?
SUCLA2 related DNA depletion syndrome follows a pattern of autosomal recessive inheritance
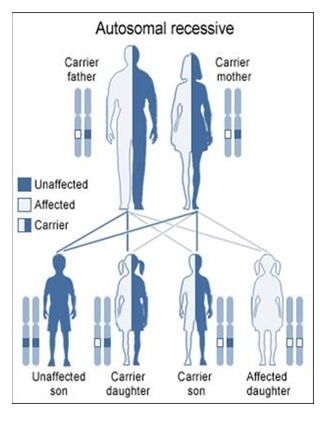
Biological Basis of the Disease
- Most commonly, the cause is a mutation to SUCLA2 gene on chromosome 13
- SUCLA2 is responsible for making an enzyme that is involved in the initial breakdown of food molecules once they enter the mitochondria
- A mutation makes faulty enzymes that don’t allow proper energy production
- SUCLA2 is also responsible for regular maintenance and repair of mtDNA in the mitochondria
- With dysfunctional SUCLA2 product, regular maintenance of DNA is affected and mtDNA molecules begin disintegrating if they aren’t repaired
Treatments
Like most mitochondrial diseases, no cure exists for this disease and the primary goal is to control the symptoms as much as possible to improve the quality of life.
- Physical therapy and occupational therapy to improve mobility and comfort
- Bracing or surgery for kyphosis and scoliosis
- Anti-epileptic drugs to treat epileptic seizures
Prognosis
Prognosis for the most part is poor, although it varies greatly from patient to patient. Severity of complications is what eventually determines the life expectancy.
References
DiMauro, Salvatore, and Michio Hirano. "Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like Episodes; Myopathy, Mitochondrial-Encephalopathy-Lactic Acidosis-Stroke." GeneReviews - NCBI Bookshelf. U.S National Library of Medicine, 27 Feb. 2001. Web. 5 July 2010. <http://www.ncbi.nlm.nih.gov/books/NBK1233/>.
Scaglia, Fernando. "MELAS Syndrome." eMedicine. WebMD, 10 May 2010. Web. 11 July 2010. <http://emedicine.medscape.com/article/946864-overview>.
DiMauro, Salvatore, and Michio Hirano. "Myoclonic Epilepsy Associated with Ragged Red Fibers." GeneReviews - NCBI Bookshelf. U.S National Library of Medicine, 3 June 2003. Web. 5 July 2010. <http://www.ncbi.nlm.nih.gov/books/NBK1520/>.
Thorburn, David R., and Shamima Rahman. "Mitochondrial DNA-Associated Leigh Syndrome and NARP." GeneReviews - NCBI Bookshelf. U.S National Library of Medicine, 30 Oct. 2003. Web. 6 July 2010. <http://www.ncbi.nlm.nih.gov/books/NBK1173/>.
Yu-Wai-Man, Patrick, and Patrick F. Chinnery. "Hereditary Optic Neuroretinopathy, LHON, Leber's Disease, Leber's Optic Atrophy, Leber's Optic Neuropathy." GeneReviews - NCBI Bookshelf. U.S National Library of Medicine, 26 Oct. 2000. Web. 7 July 2010. <http://www.ncbi.nlm.nih.gov/books/NBK1174/>.
DiMauro, Salvatore, and Michio Hirano. "mtDNA Deletion Syndromes. Includes: Kearns-Sayre Syndrome (KSS), Pearson Syndrome, Progressive External Ophthalmoplegia (PEO)." GeneReviews - NCBI Bookshelf. U.S National Library of Medicine, 17 Dec. 2003. Web. 7 July 2010. <http://www.ncbi.nlm.nih.gov/books/NBK1203/>.
Shoffner, John M. "MNGIE Syndrome, Mitochondrial Neurogastrointestinal Encephalopathy Syndrome, Myoneurogastrointestinal Encephalopathy Syndrome, Thymidine Phosphorylase Deficiency." GeneReviews - NCBI Bookshelf. U.S National Library of Medicine, 22 Apr. 2005. Web. 9 July 2010. <http://www.ncbi.nlm.nih.gov/books/NBK1179/>.
Boehnke C, Reuter U, Flach U, Schuh-Hofer S, Einhaupl KM, Arnold G.
High-dose riboflavin treatment is efficacious in migraine prophylaxis: an open study in a tertiary care centre. Eur J Neurol. 2004 Jul;11(7):475-7.
Kubota M, Sakakihara Y, Mori M, Yamagata T, Momoi-Yoshida M.Beneficial effect of L-arginine for stroke-like episode in MELAS. Brain Dev. 2004 Oct;26(7):481-3; discussion 480.
Robert McFarland MRCPCH, Prof Robert W Taylor PhD and Prof Douglass M Turnbull FRCP, A neurological perspective on mitochondrial disease. The lancet Neurology Volume 9, Issue 8, August 2010, Pages 829-840
Koga Y, Pvalko N, Nishioka J, Katayama K, Kakimoto N, Matsuishi T. MELAS and L-arginine therapy: pathophysiology of stroke-like episodes. Ann N Y Acad Sci. 2010 Jul;1201:104-10.
Kerr DS. Treatment of mitochondrial electron transport chain disorders: a review of clinical trials over the past decade. Mol Genet Metab. 2010 Mar;99(3):246-55. Epub 2009 Nov 26.
Shoffner JM, Wallace DC. Oxidative phosphorylation diseases and mitochondrial DNA mutations: diagnosis and treatment. Annu Rev Nutr. 1994;14:535-68.
Finsterer J. Treatment of mitochondrial disorders. European Journal of Paediatric Neurology 14. 2010 29-44.
P Chinnery, K Majamaa, D Turnbull and D Thorburn, Treatment for mitochondrial disorders, Cochrane Database Syst Rev 1 (2006) CD004426..
Online resources
http://www.ncbi.nlm.nih.gov/books/NBK1224/
Finsterer J. Treatment of Mitochondrial Disorders. Eur J Paediatr Neurol. 2010; 29-44