If a doctor suspects that someone has cystic fibrosis, he or she will carry out special tests including the following.
Sweat Testing
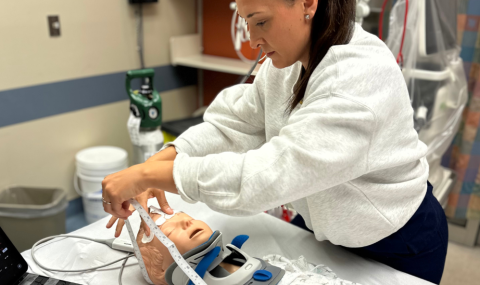
A "sweat test" is the most common test performed, in which a small area of the skin is stimulated with medication. If the sweat contains more salt than normal, it supports a diagnosis of CF.
A sweat test measures the amount of salt chemicals (sodium and chloride) in sweat. It is done to help diagnose cystic fibrosis. Normally, sweat on the skin surface contains very little sodium and chloride. People with cystic fibrosis have two to five times the normal amount of sodium and chloride in their sweat.
During the sweat test, medicine that causes a person to sweat is applied to the skin (usually on the arm or thigh). The sweat is then collected on a paper or a gauze pad, and the amount of salt chemicals in the paper or gauze is measured in a lab. Generally, chloride (sweat chloride) is measured.
A sweat test is done on any baby suspected of having cystic fibrosis. An initial test may be done as early as 48 hours of age. However, a sweat test done during the first month of life may need to be repeated. Younger babies may not produce enough sweat to give reliable test results. Also, younger babies may naturally have lower sweat chloride levels than older babies and children with cystic fibrosis.
Genetic Testing
Genetic tests, based on a blood sample can help confirm a diagnosis of CF, but these tests are more often used to determine if a person is a carrier. The standard test to diagnose CF is the sweat test, which measures the concentration of salt in the sweat. People with CF have saltier sweat than most people. Also, if someone is diagnosed with CF through a sweat test, a genetic test can show the mutations that the person is carrying.
Other Investigations
In cases other than newborn screening the physician will often discuss the history of the patient’s digestive and breathing difficulties, infections and failure to gain weight. A chest X-ray is often taken to learn whether there have been any changes in the lungs. A fecal elastase test for the presence of enzymes in the intestine may also be performed.